
 Gabe
GabeIntroducción
La dermatomiositis y la polimiositis son miopatías inflamatorias idiopáticas, caracterizadas por las características compartidas de debilidad del músculo esquelético proximal y evidencia de inflamación muscular. La DM, a diferencia de la PM, se asocia con una variedad de manifestaciones cutáneas características.
Una forma de DM, denominada DM clínicamente amiopática (También conocida como dermatomiositis sin miositis), es una afección en la que los pacientes tienen hallazgos característicos de DM en la piel sin debilidad muscular objetiva.
Existe una creciente comprensión de que el término PM está cayendo en desuso dada la mayor comprensión de que muchos casos que anteriormente se habrían clasificado como PM ahora se clasifican mejor como el síndrome antisintetasa (sin una erupción característica), miositis superpuesta (con otra enfermedad del tejido conectivo) o la miopatía necrotizante inmunomediada (IMNM). Si bien ahora se entiende que las características de la biopsia muscular son distintas en IMNM, estudios anteriores a menudo etiquetaron estos casos como PM, asumiendo que la inflamación primaria se había perdido en el muestreo de biopsia muscular.
¿Cuándo sospechar Dermatomiositis y polimiositis?
El diagnóstico de dermatomiositis (DM) o polimiositis (PM) debe sospecharse en pacientes que presentan debilidad muscular proximal. La sospecha de DM en particular debe aumentarse aún más si el paciente tiene una erupción cutánea sugestiva de DM. Tanto la PM como la DM a menudo tienen creatina quinasa (CK) elevada en la presentación, lo que también puede aumentar la sospecha de la enfermedad en un paciente que presenta debilidad que conduce a dificultades en las actividades de la vida diaria.
El diagnóstico de DM clínicamente amiopática (CADM) debe sospecharse en un paciente que presenta una erupción cutánea característica en ausencia de debilidad muscular, aunque algunos pacientes pueden tener anomalías leves en las enzimas derivadas del músculo, en la electromiografía (EMG), resonancia magnética muscular (MRI) o biopsia muscular. En particular, a pesar de carecer de una participación muscular clínicamente significativa, estos pacientes pueden tener enfermedad sistémica asociada, incluida la enfermedad pulmonar intersticial o la neoplasia maligna.
Evaluación inicial
En pacientes con sospecha de DM o PM, la presentación inicial generalmente determina la naturaleza y el alcance de las pruebas que pueden ser necesarias para confirmar el diagnóstico y excluir otros trastornos que pueden causar debilidad similar, erupciones cutáneas o enfermedad multisistémica.
Historia
Los pacientes deben ser interrogados con respecto a la duración, el modo de inicio, la ubicación y la gravedad de la debilidad y / o erupciones cutáneas. Se debe preguntar al paciente sobre su capacidad para llevar a cabo diversas actividades que comúnmente realiza, como subir escaleras, levantarse de una silla y cargar comestibles pesados u otros objetos. Evaluar la gravedad y distribución de la mialgia, así como la presencia o ausencia de dolor muscular o articular significativo, hinchazón o rigidez.
Preguntar a los pacientes sobre antecedentes de disfagia, que puede sugerir afectación esofágica, y de tos o dificultad para respirar, que puede ocurrir debido a la afectación pulmonar. Se deben buscar síntomas de otras enfermedades reumáticas sistémicas, particularmente lupus eritematoso sistémico (LES) y esclerosis sistémica (SSc), que puedan identificar un síndrome de superposición, y se debe consultar a los pacientes sobre erupciones cutáneas, fotosensibilidad, prurito, fiebre y fenómeno de Raynaud. También se debe consultar a los pacientes si han estado experimentando algún síntoma que sugiera malignidad y qué pruebas de detección y otras pruebas se han realizado para la detección del cáncer.
Exploración física
El examen de la piel, los músculos y las articulaciones es central. Los hallazgos característicos en el examen de la piel deben identificarse, si están presentes, pero a veces son sutiles. Se debe realizar un examen exhaustivo de la piel con especial atención al cuero cabelludo, la cara, los párpados, las manos, los pies, las superficies extensoras, la parte superior del pecho y la espalda, los brazos, los muslos laterales y las articulaciones. También se debe realizar un examen del pliegue ungueal, evaluando el eritema periungual, así como las anomalías capilares del pliegue ungueal. Se debe realizar un examen físico general, con especial atención al corazón y los pulmones, que deben ser cuidadosamente auscultados para detectar crepitaciones pulmonares. Se debe realizar un examen conjunto completo para detectar signos de artritis inflamatoria. Un examen neurológico y neuromuscular detallado es fundamental para determinar la gravedad y la distribución de la debilidad y de la sensibilidad muscular, así como la presencia o ausencia de otros hallazgos neurológicos anormales que pueden ser indicativos de una imitación de miositis clínica.
Pruebas de laboratorio e imágenes
Obtenemos las siguientes pruebas generales de laboratorio:
- CK es la enzima muscular más sensible y debe ser probada y seguida en todos los pacientes con sospecha de DM o PM. Los pacientes también pueden tener una elevación de otras enzimas derivadas del músculo, incluyendo lactato deshidrogenasa (LDH), aspartato aminotransferasa (AST) y alanina aminotransferasa (ALT).
- La aldolasa puede ser útil ocasionalmente en casos en los que la CK es normal, ya que un pequeño porcentaje de pacientes puede tener un nivel elevado de aldolasa, lo que sugiere que puede haber afectación perimisial. Sin embargo, no realizamos pruebas rutinarias de aldolasa. Las elevaciones de la aldolasa pueden sugerir fascitis, enfermedad pulmonar intersticial asociada a la enfermedad del tejido conectivo, o una miositis asociada con la enfermedad crónica de injerto contra huésped.
- Velocidad de sedimentación eritrocitaria (VSG) y/o proteína C reactiva (PCR): la PCR y/o la VSG a menudo son normales o solo ligeramente elevadas, incluso en pacientes con enfermedad muscular activa. La PCR y/o VSG marcadamente elevadas en un paciente con miopatía inflamatoria debe levantar la sospecha de una infección coexistente, una neoplasia maligna subyacente, o podría observarse en algunos casos de síndrome antisintetasa agudo/subagudo o enfermedad positiva al gen 5 (MDA5) asociado a la diferenciación anti-melanoma.
- Hormona estimulante de la tiroides (TSH).
Los autoanticuerpos específicos de la miositis (a menudo disponibles como panel), incluidos los anti-Jo-1
Los autoanticuerpos específicos de la miositis se asocian con síndromes clínicos particulares dentro del espectro de miopatía inflamatoria idiopática, que incluye DM y PM. Estas pruebas son positivas en el 45 al 85 por ciento de los pacientes; por lo tanto, un panel de miositis negativa no descarta un diagnóstico de DM o PM. Muchos expertos utilizan paneles de autoanticuerpos de miositis para determinar qué fenotipo de miositis en particular puede adaptarse mejor al paciente en lugar de hacer un diagnóstico de miositis.
Autoanticuerpos asociados a la miositis, incluidos los anticuerpos anti-Ro/SSA, anti-La/SSB, anti-Sm, anti-ribonucleoproteína (RNP), anti-PM-Scl y anti-Ku
Los autoanticuerpos asociados a la miositis son los que se encuentran en pacientes con otras enfermedades reumáticas sistémicas que pueden estar asociadas con la miositis.
Anticuerpos antinucleares (ANA)
Si bien el ANA detectado por los métodos estándar de inmunofluorescencia puede estar presente en hasta el 60 por ciento de los pacientes con DM o PM, las pruebas de ANA no son diagnósticas para DM o PM. Un patrón de ANA anticitoplasmático es típico tanto de los autoanticuerpos anti-MDA5 como de todos los antianticuerpos antisintetasa.
Principales patrones de enfermedad
Pacientes con hallazgos clínicos y de laboratorio característicos que son patognomónicos de la DM, que incluyen principalmente debilidad muscular proximal simétrica con hallazgos cutáneos «específicos» o patognomónicos (por ejemplo, pápulas de Gottron y / o una erupción heliotropa) en el contexto de una marcada elevación de las enzimas musculares, en quienes falta evidencia que sugiera un diagnóstico alternativo, es posible que no se requieran pruebas adicionales para confirmar el diagnóstico, incluida una biopsia de piel o músculo. Sin embargo, algunos expertos también obtienen un EMG y / o MRI en este contexto para documentar aún más que los hallazgos característicos de una miopatía inflamatoria están presentes. Otros obtienen una biopsia de piel para confirmar cambios histopatológicos compatibles con un diagnóstico de DM.

Eritema violáceo en los párpados superiores en un paciente con dermatomiositis. El eritema facial medio que no perdona los pliegues nasolabiales también está presente.
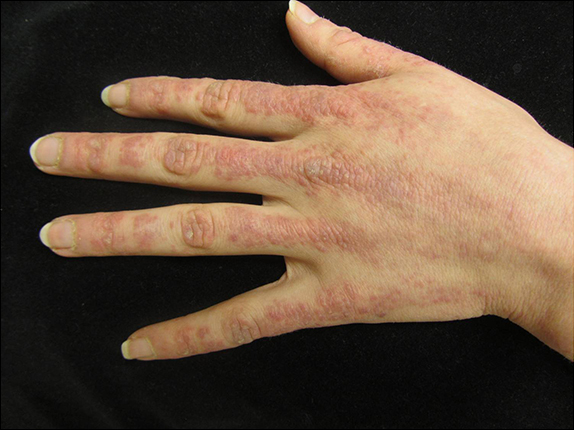
Pápulas eritematosas a violáceas en las articulaciones metacarpofalángicas y las articulaciones interfalángicas en un paciente con dermatomiositis. La erupción se extiende sobre la piel entre las articulaciones.
Pacientes con debilidad muscular sin hallazgos característicos de la piel
Los pacientes que presentan debilidad muscular proximal simétrica y enzimas musculares elevadas sin hallazgos en la piel generalmente requieren una biopsia muscular para demostrar los hallazgos histopatológicos típicos de PM o un diagnóstico alternativo como distrofia muscular, miopatía metabólica, miopatía necrosante o IBM.
EMG y / o resonancia magnética muscular se pueden realizar para identificar un sitio para la biopsia muscular en algunos casos. Dada la alta sensibilidad y especificidad de los autoanticuerpos anti-hidroximetilglutaril (HMG) coenzima A reductasa (HMGCR), los pacientes con estos anticuerpos y un fenotipo clínicamente consistente, especialmente con la exposición previa a estatinas, generalmente no necesitan someterse a una biopsia muscular.
En pacientes con debilidad muscular que es atípica (es decir, asimétrica o más distal), en los que puede ser difícil distinguir entre un trastorno miopático y neuropático que causa debilidad, se debe realizar una EMG y / o RESONANCIA magnética antes de una biopsia muscular. Si la miopatía es sugerida por EMG o MRI, se debe realizar una biopsia muscular.
Pacientes con características cutáneas sin debilidad
Un grupo distinto de pacientes con «dermatomiositis clínicamente amiopática» (CADM), históricamente llamada «dermatomiositis sine miositis», tienen hallazgos cutáneos clásicos de DM sin evidencia clínica de debilidad muscular. Hay dos subconjuntos dentro de este grupo. Un subconjunto, conocido como «dermatomiositis hipomiopática» (HDM), tiene evidencia sutil de miositis tras la investigación por laboratorio, EMG, biopsia muscular o imágenes a pesar de la falta de debilidad muscular clínica; el otro subconjunto, clasificado como «dermatomiositis amiopática» (ADM), incluye pacientes sin debilidad clínica o anomalías de laboratorio o de estudio muscular.
Pruebas de miopatía
El enfoque de las pruebas adicionales para la miopatía depende de la necesidad de una mayor certeza diagnóstica. Por ejemplo, en pacientes con hallazgos cutáneos característicos de DM, debilidad muscular proximal y CK elevada, tales pruebas pueden no ser necesarias en ausencia de un posible diagnóstico alternativo. Sin embargo, para otros pacientes en los que hay más incertidumbre, a menudo se requieren pruebas adicionales.
La EMG y los estudios de conducción nerviosa (NCS) son más útiles al principio de la evaluación de un paciente que presenta debilidad para determinar si la debilidad se debe a miopatía generalizada versus enfermedad de la neurona motora o alguna forma de neuropatía. Por lo tanto, es más útil en pacientes con debilidad muscular asimétrica o distal. Sin embargo, si una CK es marcadamente anormal (por ejemplo, mayor de 2000 unidades / L), es posible que no se requiera un EMG para confirmar la presencia de una enfermedad muscular en lugar de un trastorno neuropático. También se puede realizar un EMG para ayudar a identificar un músculo apropiado para los pacientes en los que hay dificultad para localizar un grupo muscular apropiado para la biopsia.
Electromiografía
Las anomalías características de la EMG pueden apoyar un diagnóstico de miopatía inflamatoria. Sin embargo, tales cambios no son diagnósticos de DM o PM, con hallazgos similares que ocurren en varias miopatías infecciosas, tóxicas o metabólicas. La EMG es más útil para distinguir las causas miopáticas de debilidad de los trastornos neuropáticos (por ejemplo, esclerosis lateral amiotrófica [ELA], polineuropatía periférica) y miastenia gravis, si uno no está seguro de la causa de la debilidad después de una historia y un examen cuidadosos. En pacientes con debilidad muscular equívoca en el examen, la EMG puede ser útil para identificar un músculo adecuado para la biopsia.
Imágenes del músculo esquelético
Las imágenes del músculo esquelético con resonancia magnética son una modalidad sensible no invasiva para evaluar a los pacientes con miopatía. La resonancia magnética puede demostrar áreas de edema (que es consistente pero no diagnóstico de inflamación muscular), atrofia, reemplazo graso y calcificación. A diferencia de la biopsia muscular, la resonancia magnética puede evaluar grandes áreas de músculo (por ejemplo, ambos muslos), evitando así problemas con el error de muestreo. Sin embargo, es inespecífico y puede no distinguir los cambios de la miopatía inflamatoria de los que ocurren en la rabdomiólisis, la distrofia muscular o la miopatía metabólica.
Biopsia muscular: las características histológicas de la DM y la PM incluyen necrosis de la fibra muscular, degeneración, regeneración y un infiltrado de células inflamatorias. Ciertos hallazgos característicos en estas dos enfermedades diferentes ayudan a distinguir los trastornos entre sí y reflejan sus distintas vías fisiopatológicas.
Patología muscular de la DM
La DM se caracteriza histopatológicamente por lo siguiente:
- Hay evidencia de lesión en capilares y miofibras perifasciculares. La atrofia perifascicular y la fibrosis son características, pero pueden no encontrarse en algunos pacientes biopsiados temprano en el curso de su enfermedad. En raras ocasiones, las fibras musculares anormales se agrupan en una porción del fascículo, lo que sugiere un microinfarto mediado por la disfunción de los vasos sanguíneos.
- El infiltrado inflamatorio predominante se encuentra en la región perimisial (alrededor de los haces musculares) e incluye células CD4 +, muchas de las cuales (30 a 90 por ciento) son células dendríticas plasmocitoides en lugar de células T, y también incluye macrófagos y células B. A diferencia de PM e IBM, la invasión de fibras no necróticas no es prominente.
- La expresión de la proteína A (MxA) de resistencia a los mixovirus, una proteína inducible por interferón tipo 1, en el sarcoplasma de las fibras musculares y los capilares en la expresión de inmunotinción es la anomalía histopatológica más sensible observada en la DM.
- El complejo de ataque de membrana C5b-9 del complemento terminal es detectable en las paredes de los vasos antes de la aparición de infiltración de células inflamatorias en DM pero no en PM.
Patología muscular PM
PM se caracteriza histopatológicamente por lo siguiente:
- El infiltrado celular es predominantemente endomisial (dentro de los fascículos musculares) con células inflamatorias que invaden las fibras musculares individuales. A diferencia de la DM, las fibras musculares necróticas y regeneradoras anormales se dispersan por todo el fascículo y no se limitan a una porción o área perifascicular. El tamaño de la fibra muscular es variable. No hay signos de vasculopatía o deposición de complejo inmune.
- La lesión por miofibra parece estar mediada directamente por linfocitos T citotóxicos CD8+ que rodean e invaden las miofiberes. Los macrófagos y las células dendríticas mieloides generalmente están presentes, y en algunos pacientes, también se ven células plasmáticas. El infiltrado inflamatorio puede involucrar fibras musculares no necróticas, pero este hallazgo no siempre está presente. Si bien el infiltrado es a menudo endomisial, se pueden observar infiltrados perivasculares y perimisiales. La inflamación suele estar presente, pero estos hallazgos son inespecíficos y pueden ocurrir en distrofias musculares, miopatías metabólicas después de la rabdomiólisis e IBM.
- Existe una mayor expresión de antígenos del complejo de histocompatibilidad mayor (MHC) de clase I por las fibras musculares.
- Es importante destacar que las características anteriores también son evidentes en las biopsias musculares en IBM, que es mucho más común que PM. Hasta el 20 al 30 por ciento de cualquier biopsia muscular dada en IBM puede carecer de las vacuolas con borde canónico y las inclusiones filamentosas en el microscopio electrónico.
Enlace interno
Reumatología. Medical & Gabeents
Enlace externo



