
 Gabe
GabeEl síndrome de Marfan uno de los trastornos hereditarios más comunes del tejido conectivo, predominantemente autosómica dominante con una incidencia reportada de 1 en 3000 a 5000 individuos.
Existe una amplia gama de gravedad clínica asociada con el síndrome de Marfan y trastornos relacionados, que van desde características aisladas hasta la presentación neonatal de enfermedad grave y rápidamente progresiva que involucra múltiples sistemas de órganos. Aunque muchos médicos ven el trastorno en términos de anomalías clásicas oculares, cardiovasculares y musculoesqueléticas, las manifestaciones también incluyen afectación de los pulmones, la piel y el sistema nervioso central.
Genética
Es causado por una variedad de mutaciones en el gen fibrilina 1 (FBN1). Mutaciones de FBN1 se han identificado en más del 90 % de los pacientes con síndrome de Marfan.
Sin embargo, algunos pacientes con mutaciones del gen FBN1 no tienen MFS y, en cambio, tienen un trastorno relacionado, como el síndrome de ectopia lentis u otras enfermedades, como el síndrome de Shprintzen-Goldberg, el síndrome de Weill-Marchesani o el síndrome de piel rígida.
Ausencia de mutación definida en FBN1
Alrededor del 10 por ciento de las personas con sospecha de síndrome de Marfan no tienen una mutación definida en FBN1. Algunas de estas personas pueden tener mutaciones en TGFBR1 o TGFBR2. Las mutaciones TGFBR1/TGFBR2 suelen causar el síndrome de Loeys-Dietz, con informes raros en asociación con el síndrome de aneurisma aórtico torácico familiar.
Manifestaciones clínicas
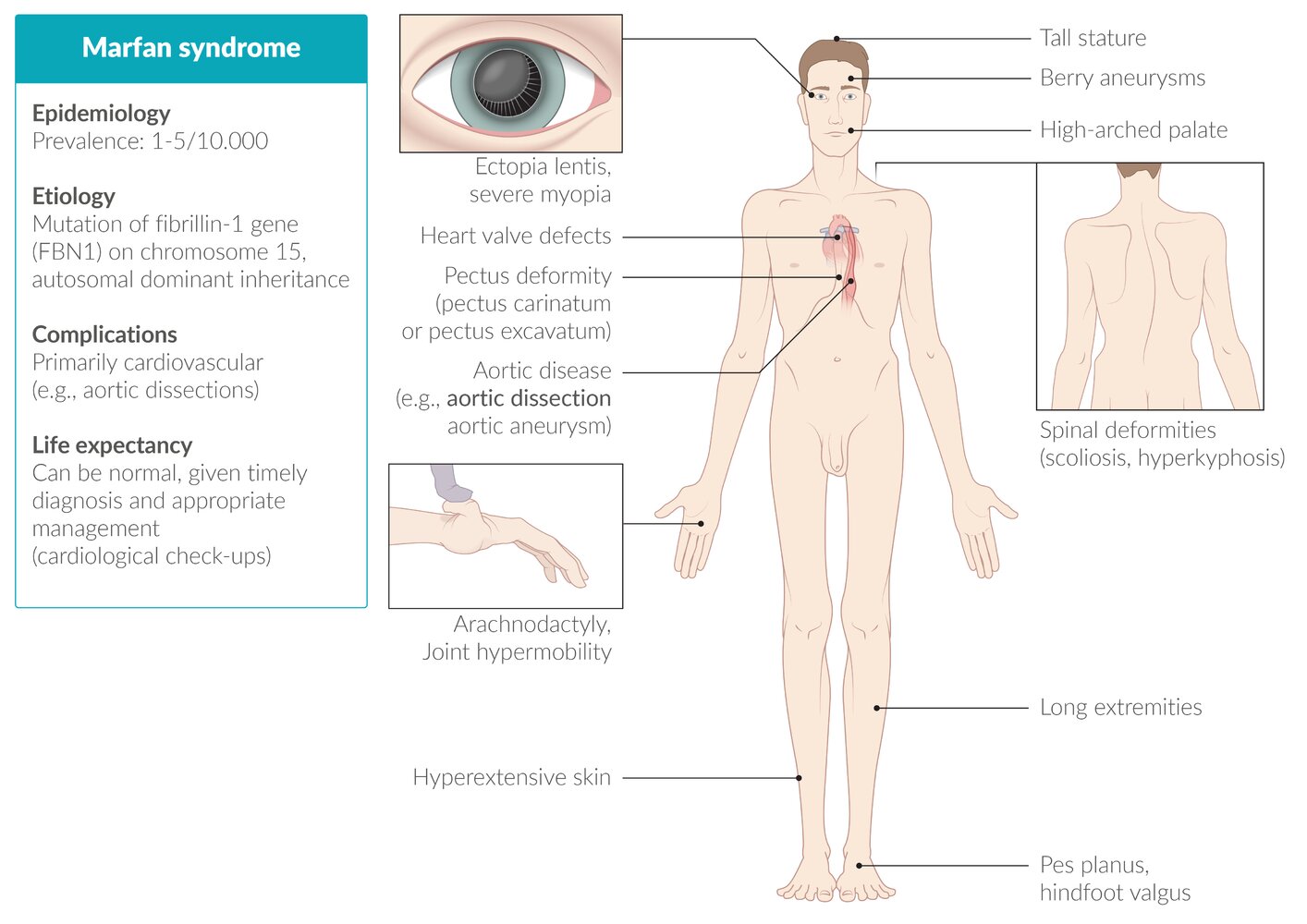
El síndrome de Marfan se caracteriza por la aparición de una serie de datos físicos característicos que afectan a diversos órganos o sistemas.
Afección cardiovascular.
- Prolapso de la válvula mitral y regurgitación. Posiblemente cause la insuficiencia mitral, que supone una complicación severa en pacientes jóvenes.
- Dilatación ventricular izquierda.
- Dilatación de arteria pulmonar.
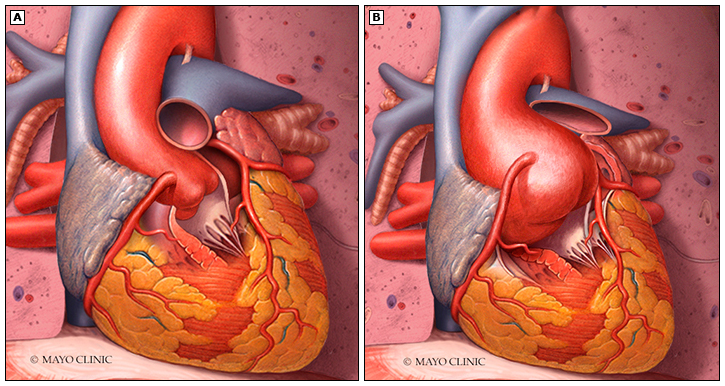
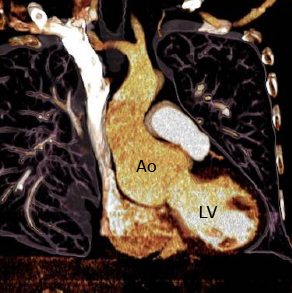
- Dilatación de la raíz de la aorta, que adquiere la forma típica de una cebolla. Esta dilatación se asocia normalmente a una incompetencia de la válvula aórtica. Constituye la principal causa de muerte, aunque ha aumentado la esperanza de vida de los pacientes con los tratamientos aplicados (de una media de edad al fallecimiento de 32±16 años en 1972 a 45±17 años en 1998).
Afectación ocular.
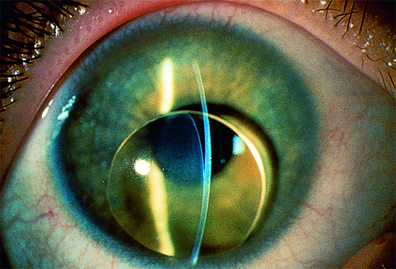
- Bilateral, subluxación del cristalino (40–56%).
- Miopía (28%).
- Desprendimiento de retina (0,78%).
Afección del aparato musculoesquelético.
- Excesivo crecimiento de las extremidades (dolicoestenomelia), es la anormalidad fenotípica fundamental, que se acentúa porque no hay aumento paralelo de la grasa ni de la masa muscular.
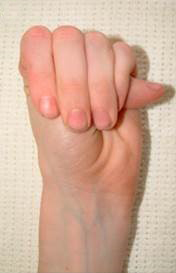
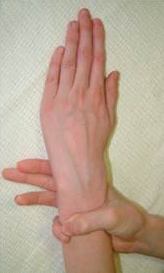
- La apariencia de los afectados de síndrome de Marfan es muy caracterísitica: muy alto, gran envergadura, muy delgado, deformidad de tronco, y dedos desproporcionadamente largos y delgados (aracnodactilia), reconocible mediante el signo de Walker-Murdoch, en el que, debido a estos dedos excesivasmente largos y la delgadez del antebrazo, el dedo pulgar y el meñique se cruzan al abarcar la muñeca contralateral.
- Disminución mineral ósea en columna vertebral y cadera, pero no se ha observado aumento de fracturas.
- Hiperlaxitud articular afecta al 85% de los pacientes menores de 18 años y el 56% de los adultos; estos pacientes sufren artralgias, mialgias y lesiones ligamentosas.
- Afección de la columna, con escoliosis muy marcada, que afecta al 60% de los pacientes.
- Protrusión acetabular en el 40% de los pacientes.
- Afección de caja torácica, con aparición de deformidad hacia fuera (pectus carinatum) o hacia dentro (pectus excavatum).
Afección del aparato respiratorio
- Puede aparecer un patrón ventilatorio restrictivo en pacientes con pectus excavatum severo que ocurre en 2/3 de los pacientes con síndrome de Marfan.
- Un 4–11% de los pacientes pueden sufrir neumotórax espontáneo.
- Pacientes adultos con síndrome de Marfan tienen una tendencia aumentada al colapso de las vías aéreas durante el sueño, lo que causa las conocidas apneas obstructivas del sueño, que pueden favorecer la somnolencia diurna.
Afección de la boca
- Paladar arqueado; la mayoría de las veces faltan los pilares del velo del paladar y hay apiñamiento de los dientes que produce mala oclusión.
Afección del sistema nervioso central
- Ectasia de la duramadre, la cual puede reducir los efectos de la anestesia epidural y ha sido relacionada a hipotensión intracraneal con cefalea asociada en algunos casos informados.
Síndrome de Marfan y embarazo
- En el embarazo el riesgo de disección aórtica, aproximadamente un 4,5% de los embarazos, está aumentado debido a la inhibición del colágeno y la elastina por estrógenos y el estado circulatorio de hipervolemia.
Diagnóstico
La presencia de una probable mutación causal de FBN1 en combinación con ciertas características clínicas brinda un fuerte respaldo al diagnóstico. El diagnóstico en casos familiares y esporádicos se basa en la presencia de manifestaciones características, particularmente dilatación/disección de la raíz aórtica y ectopia del cristalino, así como otras características sistémicas que incluyen hallazgos esqueléticos, prolapso de la válvula mitral, ectasia dural, neumotórax y estrías cutáneas.
Identificación de la dilatación aórtica
La puntuación Z de la raíz aórtica se utiliza para identificar la dilatación aórtica, ya que el tamaño de la aorta varía con el tamaño del cuerpo. Sin embargo, el uso de puntajes Z puede subestimar el tamaño de la aorta, particularmente en individuos con una gran área de superficie corporal.
Individuos jóvenes
La aplicación de criterios de diagnóstico a individuos < 20 años requiere un cuidado especial, ya que es posible que algunas características clínicas aún no hayan surgido.
Diagnóstico diferencial
El diagnóstico diferencial incluye una variedad de condiciones con características fenotípicas que se superponen parcialmente con el fenotipo de Marfan, incluidos los trastornos asociados con las mutaciones FBN1/2 o TGFBR1/2, así como una variedad de otros trastornos genéticos.
Detección de familiares
Los familiares de primer grado de pacientes con una mutación genética asociada con aneurismas aórticos y/o disección (p. ej., FBN1, TGFBR1, TGFBR2, COL3A1, ACTA2, MYH11) deben someterse a asesoramiento y pruebas genéticas. Aquellos que tengan la mutación genética deben someterse a imágenes de la aorta.
Pacientes con enfermedad aórtica
Para pacientes con aneurisma aórtico y/o disección sin una mutación conocida, se recomiendan imágenes aórticas para familiares de primer grado para identificar a aquellos con enfermedad asintomática. Si se encuentra que uno o más familiares de primer grado tienen dilatación, aneurisma o disección de la aorta torácica, entonces es razonable realizar estudios de imagen de los familiares de segundo grado.
Referencia
Summary of Diagnostic Criteria | MARFAN FOUNDATION
Valderrama Zurián FJ, Martín Gutiérrez V, Sorlí JV, Mingarro Castillo M, Ejarque Doménech I, Ortiz Uriarte R, García Ribes M. Síndrome de Marfan [Marfan’s syndrome]. Aten Primaria. 2009 May;41(5):281-4. Spanish. doi: 10.1016/j.aprim.2008.07.015. Epub 2009 May 5. PMID: 19406514; PMCID: PMC7021978.
Enlace interno



