 Gabe
GabeLa osteogénesis imperfecta (OI) es un trastorno genético del tejido conectivo caracterizado por fragilidad ósea, deformidades esqueléticas y otras manifestaciones sistémicas. Es causada principalmente por mutaciones en los genes COL1A1 y COL1A2, que codifican el colágeno tipo I. Su presentación clínica es variable, desde formas letales hasta manifestaciones leves con fracturas ocasionales.
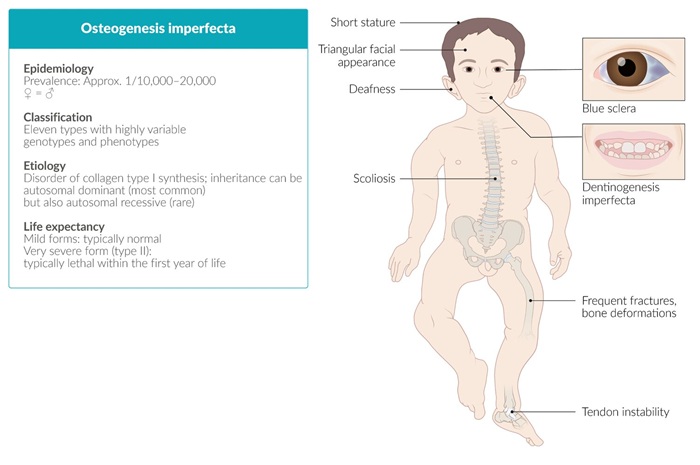
Clasificación de la Osteogénesis Imperfecta
La clasificación de Sillence divide la OI en varios tipos, con base en la severidad y características clínicas:
- Tipo I: Leve, con fragilidad ósea y escleróticas azuladas.
- Tipo II: Forma perinatal letal con fracturas múltiples in útero.
- Tipo III: Severa, con deformidades óseas progresivas y talla baja.
- Tipo IV: Moderada, con escleróticas normales y deformidades menos graves.
Manifestaciones Clínicas
- Esqueléticas: Fracturas frecuentes, deformidades óseas, baja densidad mineral ósea.
- Dentales: Dentinogénesis imperfecta.
- Auditivas: Hipoacusia en la adultez.
- Otras: Hipermovilidad articular, piel fina y escleróticas azuladas.
Diagnóstico
El diagnóstico se basa en criterios clínicos, estudios de imagen y pruebas genéticas:
- Radiografías: Osteopenia, deformidades óseas, fracturas múltiples en distintas fases de consolidación.
- Densitometría ósea: Disminución de la densidad mineral ósea.
- Pruebas genéticas: Identificación de mutaciones en COL1A1 y COL1A2.
Diagnóstico Diferencial
- Raquitismo
- Displasias esqueléticas
- Abuso infantil (ante la presencia de fracturas múltiples sin explicación adecuada)
Manejo y Tratamiento
No existe cura para la OI, pero el tratamiento busca mejorar la calidad de vida y prevenir complicaciones:
- Farmacológico:
- Bifosfonatos (pamidronato, zoledronato) para mejorar la densidad ósea.
- Suplementación con calcio y vitamina D.
- Ortopédico:
- Fijación intramedular de huesos largos para prevenir fracturas y deformidades.
- Fisioterapia para mejorar fuerza y movilidad.
- Multidisciplinario:
- Rehabilitación y terapia ocupacional.
- Seguimiento con odontología y audiología.
Pronóstico
El pronóstico varía según el tipo de OI. Las formas leves permiten una vida relativamente normal, mientras que las más severas pueden asociarse con discapacidad significativa y reducción de la esperanza de vida.
Enlaces y Referencias
- National Institutes of Health (NIH): https://www.nih.gov
- Fundación de Osteogénesis Imperfecta: https://oif.org
- Rauch, F., & Glorieux, F. H. (2004). Osteogenesis imperfecta. The Lancet, 363(9418), 1377-1385.
- Shapiro, J. R., Sponseller, P. D., & Augusciak-Dunseith, S. (2010). Osteogenesis imperfecta: A translational approach to musculoskeletal disease. The New England Journal of Medicine, 362(1), 52-62.




