
 Gabriel
GabrielIntroducción
El síndrome de Behçet, también conocido como enfermedad de Behçet, se caracteriza por aftas orales recurrentes y cualquiera de varias manifestaciones sistémicas que incluyen aftas genitales, enfermedad ocular, lesiones cutáneas, afectación gastrointestinal, enfermedad neurológica, enfermedad vascular o artritis.

Se cree que la mayoría de las manifestaciones clínicas del síndrome de Behçet se deben a la vasculitis. Entre las vasculitis sistémicas, el síndrome de Behçet es notable por su capacidad para involucrar vasos sanguíneos de todos los tamaños (pequeños, medianos y grandes) tanto en el lado arterial como en el venoso de la circulación.
Epidemiología
La prevalencia es similar en hombres y mujeres en las áreas donde es más común, pero las mujeres se ven afectadas con mayor frecuencia en los informes de los Estados Unidos y el norte de Europa. Por lo general, afecta a adultos jóvenes de 20 a 40 años, pero con poca frecuencia también se observa en niños.
Manifestaciones clínicas
La característica clínica común en pacientes con síndrome de Behçet es la presencia de úlceras mucocutáneas recurrentes y generalmente dolorosas.
La gravedad es generalmente mayor en los hombres. La mayor morbilidad y mortalidad ocurre con la enfermedad ocular (que afecta hasta dos tercios de los pacientes), la enfermedad vascular (que afecta hasta un tercio de los pacientes) y la enfermedad del sistema nervioso central (que afecta al 10 al 20 por ciento de los pacientes). Las manifestaciones cutáneas y articulares son comunes. La enfermedad renal y la afectación del sistema nervioso periférico son raras en comparación con otras vasculitis.
Ulceraciones orales: la mayoría, pero no todos, los pacientes manifiestan inicialmente ulceraciones aftosas orales recurrentes (también conocidas como aftas), que son histológicamente similares a las úlceras orales comunes y la estomatitis aftosa recurrente (RAS), pero que tienden a ser más extensas y, a menudo, múltiples.
Lesiones urogenitales: la ulceración genital, la lesión más específica para el síndrome de Behçet, ocurre en el 75 por ciento o más de los pacientes con síndrome de Behçet. Las úlceras son similares en apariencia a las aftas orales y generalmente son dolorosas. Las úlceras genitales se encuentran con mayor frecuencia en el escroto en los hombres y la vulva en las mujeres.

Lesiones cutáneas: las lesiones cutáneas también ocurren en más del 75 por ciento de los pacientes con síndrome de Behçet. Las manifestaciones cutáneas varían y pueden incluir lesiones acneiformes, erupciones papulo-vesiculo-pustulosas, pseudofoliculitis, nódulos, eritema nodoso (paniculitis septal), tromboflebitis superficial, lesiones de tipo pioderma gangrenoso, lesiones similares a eritema multiforme y púrpura palpable.
Enfermedad ocular: la enfermedad ocular ocurre en el 25 al 75 por ciento de los pacientes con síndrome de Behçet, dependiendo de la población estudiada, y en la mayoría de los casos progresa a ceguera si no se trata.
Los pacientes masculinos tienen más probabilidades de contraer enfermedades oculares, con alrededor del 75 al 80 por ciento desarrollando participación, y también tienen peores resultados visuales, incluso con tratamiento.
La uveítis es a menudo la característica dominante del síndrome de Behçet. Por lo general, es bilateral y episódica, a menudo involucra todo el tracto uveal (panuveítis) y puede no resolverse completamente entre episodios. La uveítis anterior aislada es rara.

Enfermedad neurológica: la enfermedad neurológica ocurre en menos del 10 por ciento de los pacientes con síndrome de Behçet. Se observa con más frecuencia en hombres que en mujeres.
- La enfermedad parenquimatosa se subdivide en enfermedad del tronco encefálico, enfermedad multifocal (difusa) (incluida la enfermedad del tronco encefálico, cerebral o de la médula espinal), mielopatía, enfermedad cerebral (incluida la encefalopatía, hemiparesia, pérdida hemisensorial, convulsiones, disfagia y cambios mentales como psicosis y disfunción cognitiva) y neuropatía óptica.
- La enfermedad no parenquimatosa incluye trombosis venosa cerebral, síndrome de hipertensión intracraneal (pseudotumor cerebral), síndrome meníngeo agudo y accidente cerebrovascular con poca frecuencia debido a trombosis arterial, disección o aneurisma.
Enfermedad vascular: se cree que la mayoría de las manifestaciones clínicas del síndrome de Behçet se deben a la vasculitis, y Behçet es notable por su capacidad para involucrar vasos sanguíneos de todos los tamaños (pequeños, medianos y grandes) tanto en el lado arterial como en el venoso de la circulación.
Analítica de laboratorios.
No hay pruebas de laboratorio patognomónicas en el síndrome de Behçet. Los marcadores de laboratorio de inflamación, como la velocidad de sedimentación eritrocitaria (VSG) y la proteína C reactiva (PCR), pueden elevarse en asociación con una mayor actividad de la enfermedad. Otros hallazgos de laboratorio que indican una posible actividad de la enfermedad pueden incluir aumentos en la ferritina, los glóbulos blancos, los neutrófilos, las plaquetas, el cribado plaquetario, el ancho de distribución de los glóbulos rojos (RDW), la relación neutrófilos-linfocitos, la relación monocitos-linfocitos y la relación plaquetario-linfocitos.
Diagnóstico
Debido a que las úlceras aftosas orales son tan comunes en la población general y el síndrome de Behçet es tan raro, el síndrome de Behçet se diagnostica mejor en el contexto de ulceraciones aftosas recurrentes junto con manifestaciones sistémicas características. Las manifestaciones sistémicas que deben levantar sospechas para el síndrome de Behçet incluyen enfermedad ocular, especialmente hipopión, panuveítis o vasculitis retiniana; enfermedad neurológica que incluye hallazgos característicos del parénquima del sistema nervioso central; enfermedad vascular, particularmente aneurismas de la arteria pulmonar, síndrome de Budd-Chiari y trombosis venosa cerebral; y pacientes con manifestaciones patergéticas. Las ulceraciones orales también tienden a ser más frecuentes y graves en pacientes con síndrome de Behçet. Las ulceraciones genitales son más específicas y menos sensibles para la detección del síndrome de Behçet.
Criterios de clasificación
Se han desarrollado varios criterios de diagnóstico y clasificación para el síndrome de Behçet. Estos se desarrollaron para categorizar a los pacientes con fines de estudio y no se desarrollaron para diagnosticar la enfermedad en individuos.
los criterios diagnósticos del International Study Group (ISG) publicados en 1990 parecen ser relativamente sensibles y específicos.
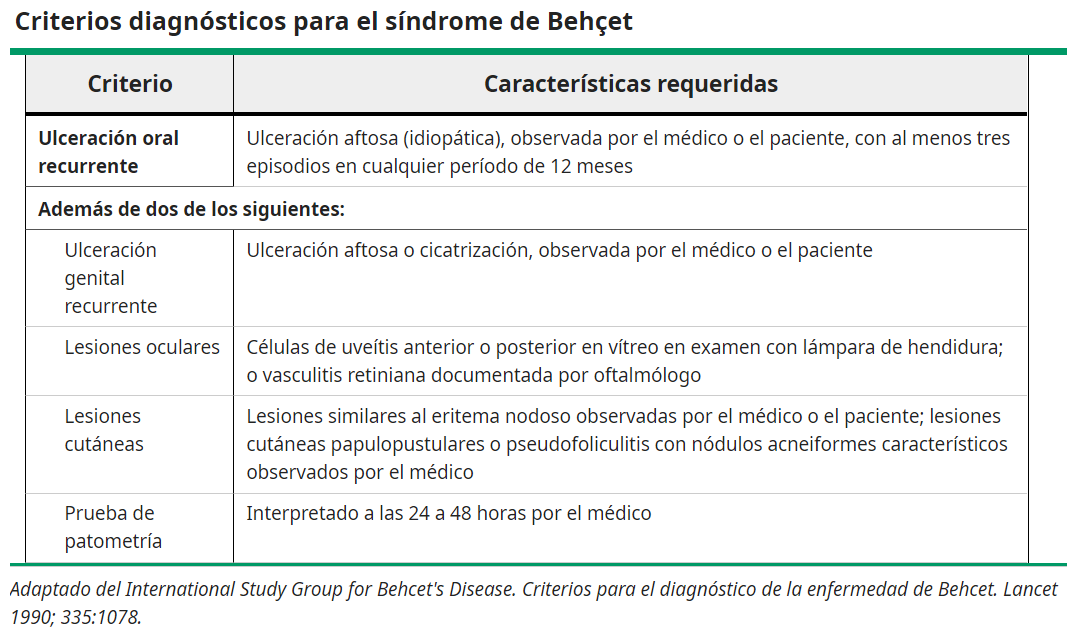
Los Criterios Internacionales para la enfermedad de Behçet (ICBD) se desarrollaron en 2006 en un esfuerzo por mejorar la sensibilidad en comparación con los criterios ISG, pero no son ampliamente aceptados.
Enlace externo



