
 Gabriel
GabrielLos eventos celulares fundamentales para el desarrollo de pancreatitis aguda incluyen:
- La señalización patológica del calcio.
- Disfunción mitocondrial.
- Activación prematura del tripsinógeno dentro de las células acinares y los macrófagos.
- Estrés del retículo endoplásmico.
- Alteración de la respuesta proteica desplegada y autofagia.
Estos eventos son provocados por toxinas comunes de las células acinares, como el alcohol, la nicotina y los ácidos biliares.
Eventos intraductales, como aumento de la presión causado por obstrucción ductal, acidificación luminal y exposición de células ductales al ácido biliar también puede desencadenar indirectamente estos eventos.
Se ha reconocido el papel mediador de la saponificación de la grasa intrapancreática, peripancreática y la isquemia condicionada por la linfa mesentérica en la gravedad de la pancreatitis aguda.
El estudio de estos mecanismos ha permitido la identificación de varios objetivos terapéuticos potenciales para futuros estudios de fármacos en pancreatitis aguda.
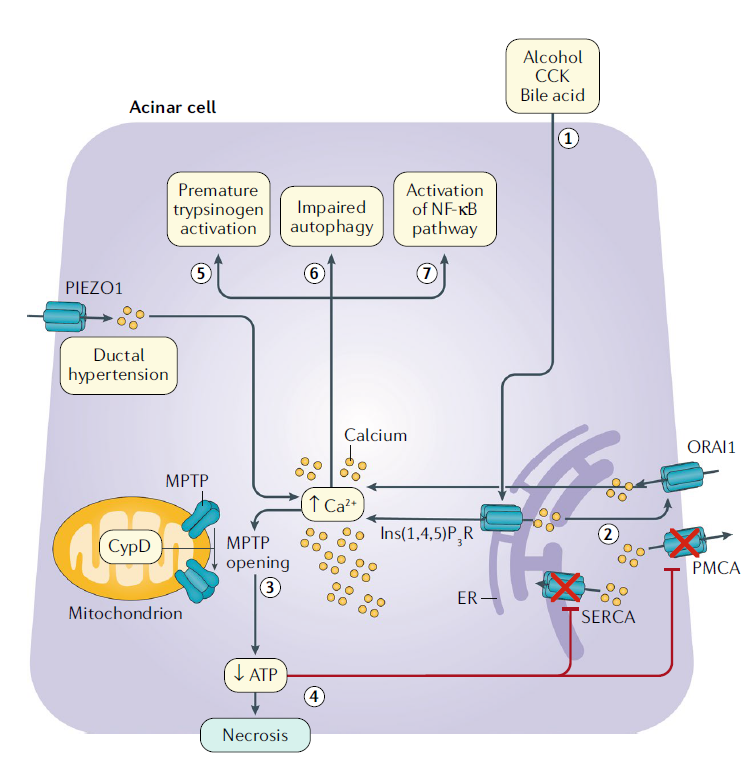
Disfunción mitocondrial mediada por calcio y muerte celular
En las células acinares, el alcohol, la colecistoquinina (CCK) y los ácidos biliares provocan la liberación de calcio mediada por el receptor de inositol 1,4,5-trifosfato desde el retículo endoplásmico.
La baja concentración de calcio resultante en el RE desencadena la apertura de la proteína 1 del canal de calcio activada por liberación de calcio, a través de la cual el calcio ingresa a la célula desde el espacio extracelular. Esto da como resultado una elevación patológica de la concentración global de calcio. La elevación del calcio da como resultado la apertura de los poros de transición de permeabilidad mitocondrial a un estado de alta conductancia, y se produce la pérdida del potencial de membrana a través de la membrana mitocondrial.
Este proceso resulta en disfunción mitocondrial y necrosis.
La disfunción mitocondrial conduce al agotamiento de ATP, lo que altera los mecanismos dependientes de ATP para reducir el calcio citosólico. Este proceso entonces acentúa y perpetúa la toxicidad patológica del calcio. La elevación patológica del calcio también causa otras vías citotóxicas, incluida la activación prematura del tripsinógeno, el deterioro de la autofagia y la activación de la vía del factor nuclear-κB. La vía NF-κB conduce a la producción de mediadores proinflamatorios.
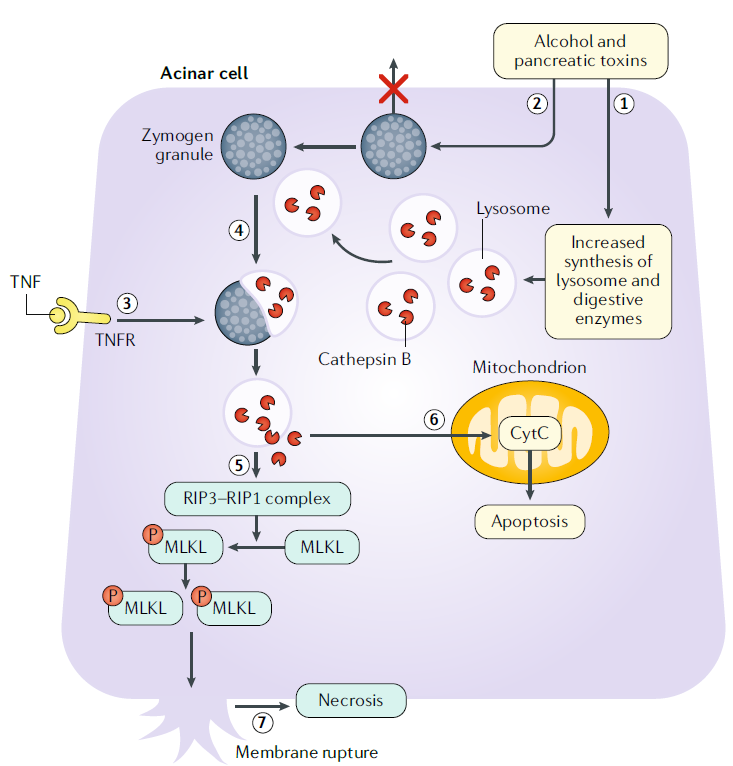
Activación prematura del tripsinógeno
En las células acinares, el alcohol y otras toxinas pancreáticas aumentan la síntesis de lisosomas y enzimas digestivas, así como también alteran la exocitosis apical de los gránulos de zimógeno en las células acinares al causar disfunción de los microtúbulos. Este proceso da como resultado la acumulación de gránulos de zimógeno.
El TNF también puede provocar la activación prematura del tripsinógeno al activar el receptor de TNF. Los eventos anteriores culminan en liberación de los lisosomas y los gránulos de zimógeno.
La catepsina B activa el tripsinógeno a tripsina. La catepsina B y la tripsina se liberan al citosol.
La catepsina B activa la proteína quinasa que interactúa con el receptor, que incluye RIP1–RIP3, y la vía similar al dominio de la quinasa de linaje mixto, que implica la oligomerización de MLKL. La liberación de proteasa intracitosólica también conduce a la activación de la caspasa 3 ejecutora de la apoptosis a través de la liberación de citocromo c de las mitocondrias.
La fosforilación y oligomerización de MLKL lo translocan a la membrana celular, provocando la ruptura de la membrana y la necrosis celular.
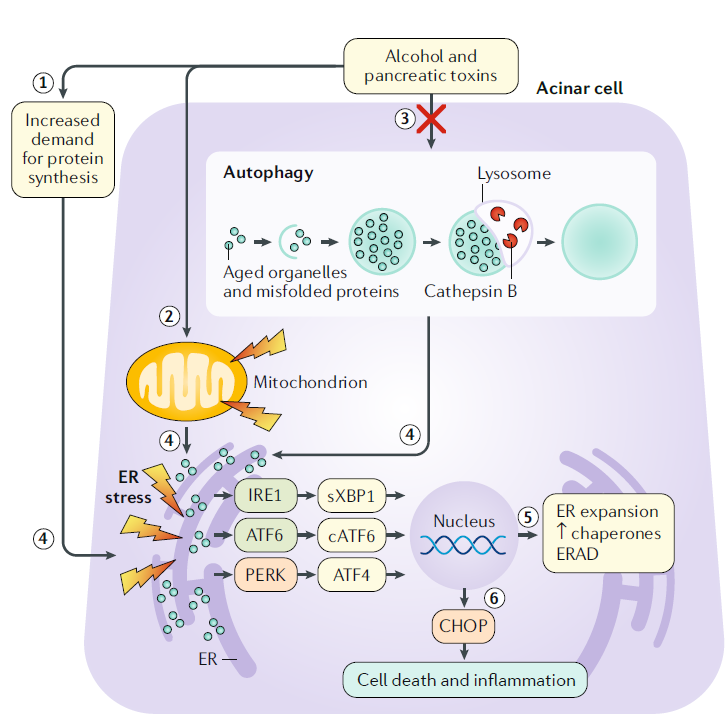
Estrés del retículo endoplásmico, respuesta de proteínas desplegadas y la autofagia
En las células acinares, las toxinas como el alcohol provocan una mayor demanda de síntesis de proteínas, disfunción mitocondrial y deterioro de la autofagia.
En condiciones fisiológicas, la autofagia procesa y recicla diversos contenidos citoplasmáticos envejecidos y defectuosos. La autofagia primero enuclea el contenido citosólico dentro de una doble membrana abierta. Los bordes de la doble membrana se unen para formar un autofagosoma, que se fusiona con un lisosoma para formar un autolisosoma. Las hidrolasas en el lisosoma luego degradan el contenido adjunto para reciclarlo. Los eventos anteriores dan como resultado estrés en el retículo endoplásmico, que ocurre cuando la demanda de síntesis de proteínas y la acumulación de proteínas mal plegadas o desplegadas en una célula abruman la capacidad para procesarlas.
Las proteínas transmembrana como la enzima 1 que requiere inositol (IRE1), el factor de transcripción activador 6 (ATF6) y la proteína quinasa quinasa ER tipo ARN (PERK) detectan proteínas mal plegadas. Los efectores de la vía IRE1 y ATF6 empalmados X-box binding protein 1 (sXBP1), ATF6 y ATF6 escindido (cATF6) son factores de transcripción para genes implicados en la expansión del retículo endoplásmico, procesos de chaperonas moleculares y degradación, para satisfacer las demandas metabólicas y de síntesis de proteínas de la célula.
Bajo estrés extremo del retículo endoplásmico, la vía PERK de la respuesta proteica desplegada (UPR, por sus siglas en inglés) da como resultado apoptosis e inflamación mediada por la proteína homóloga de CEBP.
Información adicional
Aunque una serie de condiciones pueden precipitar la pancreatitis aguda, sólo una pequeña fracción de los pacientes con estas condiciones predisponentes desarrolla pancreatitis aguda. Por ejemplo, la incidencia de pancreatitis aguda es sólo del 3 al 7 % en pacientes con cálculos biliares y del 10 % en alcohólicos.
No está claro por qué la pancreatitis inducida por el alcohol ocurre solo después de muchos años de abuso de alcohol y no después de un solo atracón en personas que no están habituadas al consumo de alcohol. Sin embargo, se han propuesto varios mecanismos:
- Sensibilización de células acinares a la activación prematura de zimógenos inducida por colecistoquinina (CCK).
- Potenciación del efecto de CCK sobre la activación de factores de transcripción, factor nuclear kB y proteína activadora-1.
- Generación de metabolitos tóxicos como acetaldehído y ésteres etílicos de ácidos grasos.
- Sensibilización del páncreas a los efectos tóxicos del virus coxsackie B.
- Activación de las células estrelladas pancreáticas por acetaldehído y estrés oxidativo y posterior aumento de la producción de colágeno y otras proteínas de la matriz.
Se han sugerido dos factores como posible evento iniciador en la pancreatitis por cálculos biliares:
- Reflujo de bilis hacia el conducto pancreático debido a la obstrucción transitoria de la ampolla durante el paso de los cálculos biliares.
- Obstrucción en la ampolla secundaria a cálculos o edema resultante del paso de una piedra.
En la hipertrigliceridemia, los ácidos grasos libres se liberan de los triglicéridos séricos en concentraciones tóxicas por la acción de la lipasa pancreática dentro de los capilares pancreáticos.
También se ha propuesto la activación prematura de zimógenos pancreáticos dentro del páncreas como mecanismo patogénico de los ataques agudos de pancreatitis observados en pacientes con pancreatitis hereditaria.
No está claro cómo las mutaciones de CFTR podrían producir pancreatitis aguda. Una posible explicación es que las mutaciones están asociadas con la producción de un jugo pancreático más concentrado y ácido que conduce a la obstrucción ductal o a la alteración de la función de las células acinares.
Cada vez es más evidente que el requisito central para la inducción de pancreatitis aguda es la activación de enzimas proteolíticas intraacinares, lo que finalmente conduce a una lesión autodigestiva de la glándula.
Las enzimas pancreáticas activadas, el deterioro de la microcirculación y la liberación de mediadores inflamatorios conducen a un rápido empeoramiento del daño pancreático y la necrosis. Sin embargo, aproximadamente el 80 por ciento de los pacientes con pancreatitis desarrollan solo pancreatitis intersticial en lugar de pancreatitis necrosante; los factores involucrados en la limitación del daño pancreático no se conocen bien.
Algunos pacientes con daño pancreático severo desarrollan síndrome de respuesta inflamatoria sistémica (SIRS) probablemente mediado por enzimas pancreáticas activadas y citocinas liberadas a la circulación desde el páncreas inflamado. Un síndrome de respuesta antiinflamatoria compensada (CARS) equilibra el SIRS y conduce a la recuperación. Un desequilibrio entre SIRS y CARS da como resultado una falla orgánica grave con alta morbilidad y mortalidad. Las causas de tal desequilibrio no se entienden claramente.
Durante el curso de la pancreatitis aguda, la barrera intestinal se ve comprometida, lo que lleva a la translocación de bacterias, lo que puede provocar una infección local y sistémica. Las consecuencias de la translocación bacteriana desde el intestino en la pancreatitis aguda pueden ser letales. La infección bacteriana local de los tejidos pancreáticos y peripancreáticos ocurre en aproximadamente el 30 por ciento de los pacientes con pancreatitis aguda grave, lo que puede provocar una falla multiorgánica y sus secuelas.
La evidencia cuestiona el papel de la activación prematura del tripsinógeno como evento principal en la patogénesis de la pancreatitis aguda y esto puede causar solo lesión acinar pero no la respuesta inflamatoria en áreas pancreáticas y extrapancreáticas. La respuesta inflamatoria pancreática y extrapancreática (sistémica) más pronunciada está impulsada por la activación de NFkB. El retículo endoplásmico y el estrés oxidativo junto con la inducción de una vía autofágica defectuosa son otros factores importantes descritos en la patogenia junto con el papel de los TLR4 (Toll-Like Receptor family).
Fuente
Lee, P.J., Papachristou, G.I. New insights into acute pancreatitis. Nat Rev Gastroenterol Hepatol 16, 479–496 (2019). https://doi.org/10.1038/s41575-019-0158-2
Mederos MA, Reber HA, Girgis MD. Acute Pancreatitis: A Review. JAMA. 2021;325(4):382–390. doi:10.1001/jama.2020.20317
Enlace interno



