
 Gabriel
GabrielLa enfermedad de Wilson es un trastorno hereditario potencialmente tratable del metabolismo del cobre que se caracteriza por la acumulación patológica de cobre. Es causada por mutaciones en ATP7B, que codifica una ATPasa transmembrana que transporta cobre, lo que conduce a alteraciones de la homeostasis del cobre y sobrecarga de cobre en el hígado, el cerebro y otros órganos.
El curso clínico de la enfermedad puede variar en el tipo y la gravedad de los síntomas, pero la enfermedad hepática progresiva es una característica común. Los pacientes también pueden presentar trastornos neurológicos y síntomas psiquiátricos.
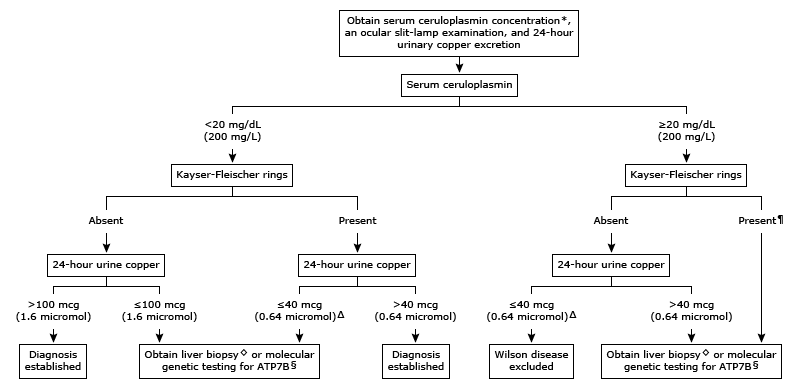
La enfermedad de Wilson se diagnostica mediante algoritmos de diagnóstico que incorporan síntomas y signos clínicos, medidas del metabolismo del cobre y análisis del ADN de ATP7B.
Los tratamientos disponibles incluyen la terapia de quelación y las sales de zinc, que revierten la sobrecarga de cobre mediante diferentes mecanismos. Además, el trasplante de hígado está indicado en casos seleccionados.
Con un diagnóstico y tratamiento tempranos, el pronóstico es bueno; sin embargo, una cuestión importante es diagnosticar a los pacientes antes de la aparición de síntomas graves. Por lo tanto, los avances en el cribado de esta enfermedad pueden traer un diagnóstico más temprano y mejoras en el tratamiento.

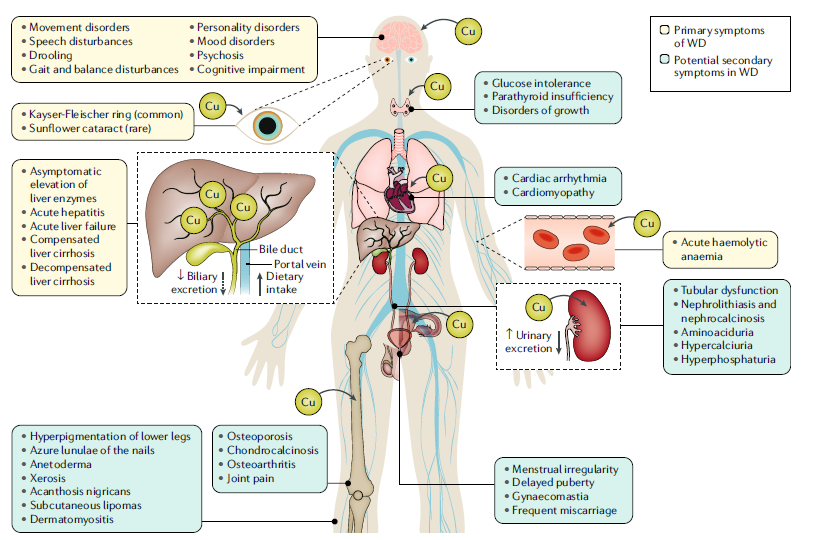
El cobre (Cu) de la dieta se transporta a través de la vena porta y se deposita en el hígado, que es el órgano central de la homeostasis sistémica del Cu. El deterioro de la excreción biliar de Cu en la enfermedad de Wilson (EW) conduce a una acumulación gradual de Cu en el hígado. Cuando la capacidad del hígado para almacenar Cu se agota, cantidades excesivas de Cu no unido a ceruloplasmina ingresan a la circulación sistémica y se depositan y acumulan en varios órganos, ejerciendo toxicidad por Cu extrahepático. El Cu se acumula en la córnea, el cerebro, los glóbulos rojos, las células del músculo esquelético y cardíaco, las membranas sinoviales de las grandes articulaciones y el parénquima renal, provocando las diversas manifestaciones clínicas. Las manifestaciones clínicas primarias están asociadas con la acumulación de Cu en el hígado, el cerebro y los ojos. El Cu plasmático no unido a ceruloplasmina se filtra por el epitelio tubular renal y se excreta por la orina.
Referencia
Członkowska, A., Litwin, T., Dusek, P. et al. Wilson disease. Nat Rev Dis Primers 4, 21 (2018). https://doi.org/10.1038/s41572-018-0018-3
Enlace interno



