
 Gabe
GabeIntroducción
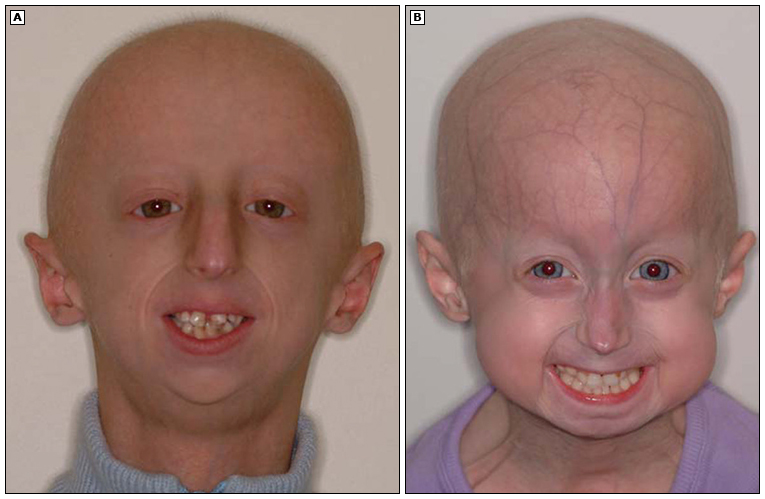
Progeria es un trastorno autosómico dominante extremadamente raro, fatal, caracterizado por un envejecimiento acelerado que comienza en la infancia. Los pacientes presentan una apariencia facial característica, alopecia, lipoatrofia, displasia esquelética y aterosclerosis que resulta en infarto de miocardio, accidente cerebrovascular y, en última instancia, la muerte en la segunda década de la vida.
Epidemiologia
El síndrome de progeria de Hutchinson-Gilford es un trastorno extremadamente raro, con una incidencia estimada de uno de cada cuatro a ocho millones de nacimientos
Patogénesis
Es causado por una sustitución de un solo nucleótido en el gen de lamina, que codifica dos tipos diferentes de isoformas de proteínas (lamina A y lamina C).
La mutación causa la producción de una proteína única de lamina A mutante llamada progerina.
La proteína progerina interrumpe la organización normal de la lámina nuclear y se plantea la hipótesis de causar sangrado nuclear, heterocromatina desorganizada y transcripción de genes desregulados. Se cree que esta inestabilidad genómica causa el envejecimiento prematuro.
Manifestaciones clínicas
Los niños afectados con síndrome de progeria de Hutchinson-Gilford parecen normales al nacer, pero las manifestaciones clínicas se hacen evidentes en los primeros años de vida. Estos incluyen la falta de crecimiento; anomalías dermatológicas, musculoesqueléticas y neurológicas; y, finalmente, enfermedades cardiovasculares que limitan la vida.
Los niños desarrollan una apariencia facial característica que incluye cianosis circumoral; ojos prominentes; nariz delgada y con pico; micrognatia; retrognatia; labios delgados; pérdida de cejas; y venas prominentes del cuero cabelludo
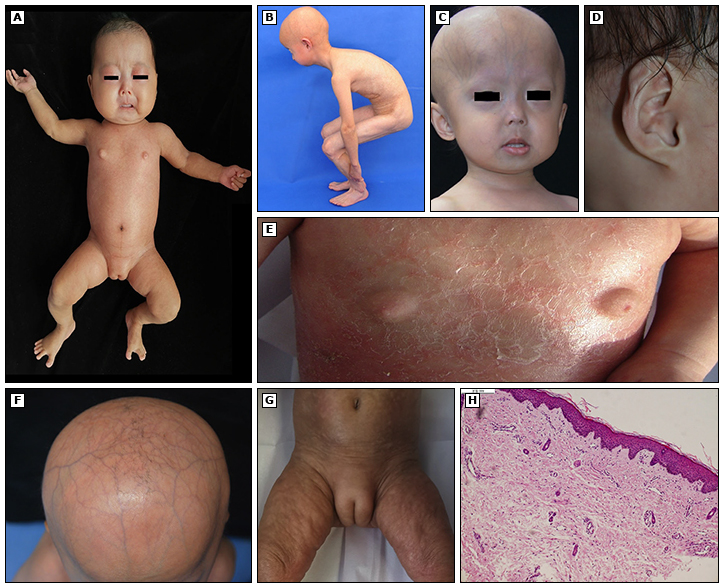
(A) Apariencia corporal general y piel esclerótica en 3 meses de edad.
(B) Rigidez articular.
(C) Características craneofaciales distintas en niños de 2 años.
(D) Lóbulo de oreja pequeño.
(E) Distintos «pezones blandos protuberantes» en 6 meses de edad.
(F) Alopecia y venas prominentes del cuero cabelludo.
(G) «Protuberant soft labium majus» en 4 meses de edad.
(H) Los hallazgos histopatológicos revelaron hiperplasia de colágeno dérmico e infiltración linfocítica perivascular (tinción H&E ×100).
Hallazgos de laboratorio
Se asocia con varias anomalías de laboratorio y de imágenes, incluyendo:
- Disminución de los niveles séricos de leptina.
- Resistencia a la insulina en hasta la mitad de los pacientes.
- Disminución de la densidad ósea.
- Hallazgos radiográficos de acro-osteolisis, reabsorción clavicular y coxa valga.
- Otros estudios de laboratorio de rutina generalmente no son notables. Se han reportado elevaciones leves en las plaquetas y tiempo prolongado de protrombina
Diagnostico
se sospecha en un niño que presenta las siguientes características:
- Incapacidad para prosperar en el primer año de vida
- Apariencia facial característica con micrognatia, ojos prominentes y cianosis circumoral
- Alopecia y venas prominentes del cuero cabelludo
- La piel esclerótica cambia con la bolsa y los hoyuelos/moteados, especialmente en el abdomen
- Disminución del rango de movimiento articular y contracturas articulares
El diagnóstico se establece en base a la presencia de las características clínicas enumeradas anteriormente y la identificación mediante pruebas genéticas de la variante patogénica causal conocida en el gen LMNA.
Medidas de soporte
implica garantizar una nutrición óptima, el monitoreo de la progresión de la enfermedad y el tratamiento de las complicaciones a medida que se presentan.
En noviembre de 2020, lonafarnib, un inhibidor oral de la farnesiltransferasa, fue aprobado por la Administración de Alimentos y Medicamentos de los Estados Unidos para el tratamiento en pacientes de 12 meses de edad en adelante.
Se plantea la hipótesis de que el lonafarnib puede inhibir la formación de la proteína progerina aberrante de lamina A y evitar su anclaje a la membrana nuclear interna, mejorando así potencialmente el estado de la enfermedad.
La principal preocupación es la aterosclerosis progresiva acelerada que conduce a infarto de miocardio, ataque isquémico transitorio y accidente cerebrovascular. El estado cardíaco y neurológico del paciente debe investigarse, incluso en pacientes asintomáticos, al menos una vez al año. La evaluación cardiovascular debe incluir electrocardiograma, medición de la presión arterial, pruebas de perfil lipídico y ecocardiografía.



