 Gabe
GabeEl monóxido de carbono (CO) es un gas inodoro, insípido, incoloro y no irritante formado por la combustión de hidrocarburos. La concentración atmosférica de CO es generalmente inferior al 0,001 por ciento, pero puede ser mayor en áreas urbanas o ambientes cerrados.
El CO se une a la hemoglobina con mucha mayor afinidad que el oxígeno, formando carboxihemoglobina (COHb) y resultando en un transporte y utilización de oxígeno deteriorado.
El CO también puede precipitar una cascada inflamatoria que resulta en peroxidación lipídica del sistema nervioso central (SNC) causando secuelas neurológicas tardías.
PATOGENIA DE LA INTOXICACIÓN POR MONÓXIDO DE CARBONO
El CO se une a la fracción de hierro del hemo (y otras porfirinas) con aproximadamente 240 veces la afinidad de la carboxihemoglobina formadora de oxígeno (COHb). Esto induce un cambio alostérico que disminuye en gran medida la capacidad de los otros tres sitios de unión al oxígeno en la hemoglobina para descargar oxígeno a los tejidos periféricos. Esto resulta en una deformación y desplazamiento hacia la izquierda de la curva de disociación de oxihemoglobina, y agrava el deterioro en el suministro de oxígeno tisular.
Aproximadamente del 10 al 15 por ciento del CO es extravascular y se une a moléculas como la mioglobina, los citocromos y la NADPH reductasa, lo que resulta en un deterioro de la fosforilación oxidativa a nivel mitocondrial. La vida media del CO unido a estas moléculas es más larga que la de COHb. La importancia de estos efectos mediados por la hemoglobina se ha documentado mejor en el corazón, donde la disfunción mitocondrial debido al CO puede producir aturdimiento miocárdico a pesar del suministro adecuado de oxígeno.
El CO produce la generación de superóxido y estrés oxidativo, lo que probablemente contribuye a la peroxidación lipídica y la lesión neurológica.
![Normal: La Hb se une al oxígeno y lo suministra al tejido periférico con bajo PO2. Citocromo c reducido (CytC) transfiere su electrón (e–) a la subunidad 1 de la citocromo c oxidasa (CytUn– centro binuclear con hemo a y cobre [CuUn]). El electrón reduce el oxígeno (O2) en la subunidad 2 (CytA3– centro binuclear con hemo a3 y cobre [CuB]), formando agua y transportando un protón (H+) a través de la membrana mitocondrial interna.
Toxicidad del CO: El CO se une competitivamente a la Hb con el O2, reduciendo la capacidad total de transporte de oxígeno al: (1) unirse preferentemente al CO en lugar de O2(efecto similar a la anemia) y (2) estabiliza el estado cuaternario relajado de Hb, que se une a O2con mayor afinidad y no lo liberará en PO bajo2medio ambiente. El CO se une competitivamente con O2en el hemo reducido a3 en la subunidad 2. Esto causa: (1) inhibición de la reducción de O2al agua (el destino final de los electrones en la cadena de transporte de electrones); (2) cese de la transferencia de H+ al espacio intermembrana, cerrando la generación de ATP a través de la ATP sintasa; y (3) acumulación de electrones que entran en la cadena de transporte de electrones a través de los complejos I y III que pueden producir superóxido que conduce a efectos nocivos.](https://gabeents.com/wp-content/uploads/2022/11/HemoglbnmitochndrlCOnorm.jpg)
Normal: La Hb se une al oxígeno y lo suministra al tejido periférico con bajo PO2. Citocromo c reducido (CytC) transfiere su electrón (e–) a la subunidad 1 de la citocromo c oxidasa (CytUn– centro binuclear con hemo a y cobre [CuUn]). El electrón reduce el oxígeno (O2) en la subunidad 2 (CytA3– centro binuclear con hemo a3 y cobre [CuB]), formando agua y transportando un protón (H+) a través de la membrana mitocondrial interna.
Toxicidad del CO: El CO se une competitivamente a la Hb con el O2, reduciendo la capacidad total de transporte de oxígeno al: (1) unirse preferentemente al CO en lugar de O2(efecto similar a la anemia) y (2) estabiliza el estado cuaternario relajado de Hb, que se une a O2con mayor afinidad y no lo liberará en PO bajo2medio ambiente. El CO se une competitivamente con O2en el hemo reducido a3 en la subunidad 2. Esto causa: (1) inhibición de la reducción de O2al agua (el destino final de los electrones en la cadena de transporte de electrones); (2) cese de la transferencia de H+ al espacio intermembrana, cerrando la generación de ATP a través de la ATP sintasa; y (3) acumulación de electrones que entran en la cadena de transporte de electrones a través de los complejos I y III que pueden producir superóxido que conduce a efectos nocivos.
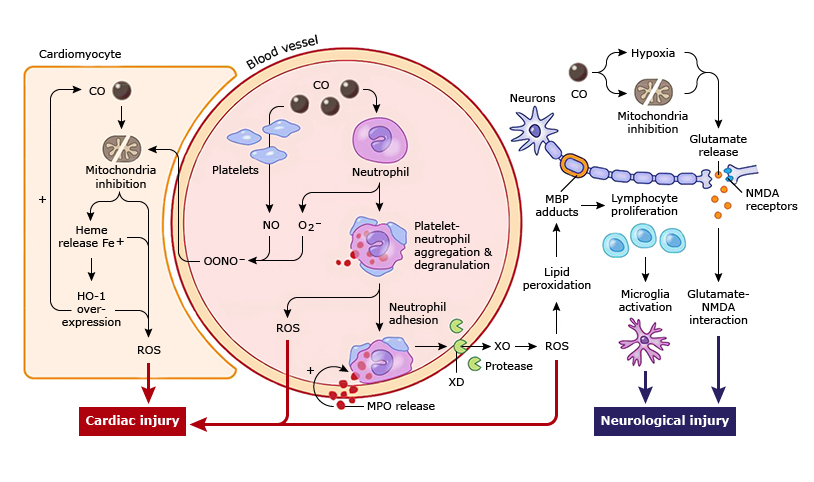
El CO activa las plaquetas desplazando el óxido nítrico plaquetario (NO) de las hemoproteínas de superficie: El NO reacciona con los radicales libres de oxígeno (O2–) para producir peroxinitrito (ONOO–), que inhibe la función mitocondrial y activa las plaquetas y los neutrófilos. La inhibición de las mitocondrias conduce a una mayor producción de especies reactivas de oxígeno (ROS) y causa la liberación de hemo libre y el consiguiente aumento de la hemo oxigenasa 1 (HO-1), causando aún más estrés oxidativo. HO-1 metaboliza el hemo libre para producir más CO endógeno, creando un circuito de retroalimentación positiva localmente. Los neutrófilos activados se desgranularán y liberarán mieloperoxidasa, causando más activación de neutrófilos y adhesión. Las proteasas liberadas por los neutrófilos pueden oxidar la xantina deshidrogenasa (XD) de las células endoteliales a la xantina oxidasa (XO), generando especies reactivas de oxígeno que causan daño celular y peroxidación lipídica, específicamente en la proteína básica de mielina (MBP). Cuando se peroxida, MBP forma aductos que causan proliferación de linfocitos, activación de microglia y, en última instancia, lesión neurológica.
Los efectos generales de la hipoxia y el efecto de la toxicidad del CO directamente sobre las mitocondrias causan la liberación de glutamato, que activa los receptores de N-metil-D-aspartato (NDMA), lo que conduce aún más a lesiones neurológicas.
DIAGNÓSTICO DE INTOXICACIÓN POR MONÓXIDO DE CARBONO
El diagnóstico de intoxicación por CO se realiza en pacientes con exposición conocida o sospechada al CO junto con un nivel elevado de carboxihemoglobina (COHb) medido por cooximetría de una muestra de gases en sangre. En pacientes hemodinámicamente estables, las muestras venosas son precisas.
La oximetría de pulso estándar (SpO)2) no puede detectar la exposición al CO, ya que no diferencia el COHb de la oxihemoglobina. Los oxímetros de pulso de ocho longitudes de onda que miden COHb y metahemoglobina están disponibles, pero no se consideran lo suficientemente precisos como para sustituir la cooximetría sanguínea, aunque pueden tener un papel como prueba de detección.
El tratamiento con hidroxocobalamina (que podría administrarse prehospitalaria por presunta exposición al cianuro) puede interferir con la medición de COHb que conduce a resultados inexactos, y se informan niveles falsamente bajos y elevados. En pacientes con posible exposición al cianuro (p. ej., llevados al hospital por un incendio), los médicos que asuman la atención deben preguntar si se administró hidroxocobalamina.
OXIGENOTERAPIA EN INTOXICACIÓN POR MONÓXIDO DE CARBONO
Las intervenciones clave en el tratamiento del paciente envenenado con CO son la eliminación rápida de la fuente de CO y el suministro de oxígeno de alto flujo.
Oxígeno de alto flujo: el oxígeno de alto flujo (100 por ciento) se inicia a través de una máscara facial que no se respira. La eliminación del CO comienza una vez que el paciente es retirado de la exposición y es casi exclusivamente a través de la circulación pulmonar a través de la unión competitiva de la hemoglobina por oxígeno.
La vida media de COHb en el aire de la sala de respiración de un paciente es de aproximadamente 250 a 320 minutos; esto disminuye a 90 minutos con oxígeno de alto flujo (>15 L / minuto) proporcionado a través de una mascarilla de alto flujo.
En pacientes que sufren de envenenamiento por CO por un incendio o inhalación de humo que están gravemente enfermos (es decir, aquellos con coma, convulsiones o compromiso hemodinámico asociado con una acidosis metabólica y aumento de la concentración de lactato en sangre), recomendamos la administración empírica dehidroxicobalamina para tratar la posible toxicidad por cianuro.
Dado que la exposición al cianuro puede ocurrir por incendios e inhalación de humo y existe una superposición en la presentación clínica con el CO, es difícil excluir definitivamente la toxicidad concomitante del cianuro, que puede afectar aún más la utilización de oxígeno en los tejidos y exacerbar la hipoxia celular.
La dosis de hidroxocobalamina es de 70 mg/kg por vía intravenosa (IV; 5 g es la dosis estándar para adultos) y puede repetirse después de 10 a 15 minutos si el paciente no tiene una mejoría clínica rápida. Los servicios de medicina de emergencia pueden haber administrado hidroxocobalamina en el camino al hospital.
Oxigenoterapia hiperbárica proporciona el mayor beneficio si el tratamiento comienza lo antes posible, idealmente dentro de las seis horas, para aumentar la eliminación de la carboxihemoglobinemia y mejorar la oxigenación del tejid.
Es importante destacar que, incluso si la COHb ha disminuido por debajo de los umbrales recomendados mencionados anteriormente, el tratamiento tardío con OHB aún puede proporcionar beneficios; sin embargo, no está probado si han pasado más de 24 horas después de la exposición al CO. Todos los pacientes seleccionados para recibir OHB deben recibir al menos un tratamiento a 2.5 a 3 atmósferas absolutas (ATA) tan pronto como sea posible para revertir los efectos agudos de la intoxicación por CO, con una posible terapia adicional dirigida a la limitación o prevención del síndrome neuropsiquiátrico tardío.
HBO expone a los pacientes al 100 por ciento de oxígeno en condiciones supraatmosféricas, lo que disminuye la vida media de COHb a 30 minutos de aproximadamente 90 minutos con oxígeno normobárico al 100 por ciento.
La cantidad de oxígeno disuelto en la sangre también aumenta de aproximadamente 0.3 a 6 ml por dL, lo que aumenta sustancialmente el suministro de oxígeno no unido a la hemoglobina a los tejidos. HBO inhibe el estallido oxidativo de neutrófilos, la producción de xantina oxidasa y la peroxidación lipídica.
SÍNDROME NEUROPSIQUIÁTRICO TARDÍO
El síndrome de secuelas neurológicas tardías (DNS) incluye grados variables de déficits cognitivos, cambios de personalidad, trastornos del movimiento y déficits neurológicos focales. El DNS ocurre en 15 a 40 por ciento de los pacientes con exposición significativa al CO.
Se informa que el DNS surge de 3 a 240 días después de la recuperación aparente, generalmente ocurre dentro de los 20 días posteriores a la intoxicación por CO. Los déficits pueden persistir durante un año o más.
Aunque el DNS se asocia con anomalías en el globo pálido y la sustancia blanca profunda en la tomografía computarizada (TC), la resonancia magnética (IRM) y la tomografía por emisión de positrones (TEP), estos hallazgos generalmente no se encuentran en la presentación aguda.
El infarto hemorrágico del globo pálido y, con menos frecuencia, de la sustancia blanca profunda se han reportado raramente después de una intoxicación aguda.
El desarrollo de DNS se correlaciona mal con los niveles de carboxihemoglobina (COHb), aunque la mayoría de los casos se asocian con pérdida de conciencia durante la intoxicación aguda.
El mecanismo de DNS no se comprende completamente, pero probablemente implica peroxidación lipídica por especies reactivas de oxígeno generadas por xantina oxidasa. La xantina oxidasa se produce in situ a partir de la xantina deshidrogenasa a través de enzimas liberadas por los glóbulos blancos que se adhieren a las células endoteliales dañadas. Durante la recuperación de la exposición al CO, los eventos análogos a la lesión por isquemia-reperfusión y la exposición a la hiperoxia pueden exacerbar el daño oxidativo inicial.
FUENTES
 Cargando…
Cargando…
Cargando…
Cargando…




