
 Gabe
GabeNeurotoxinas
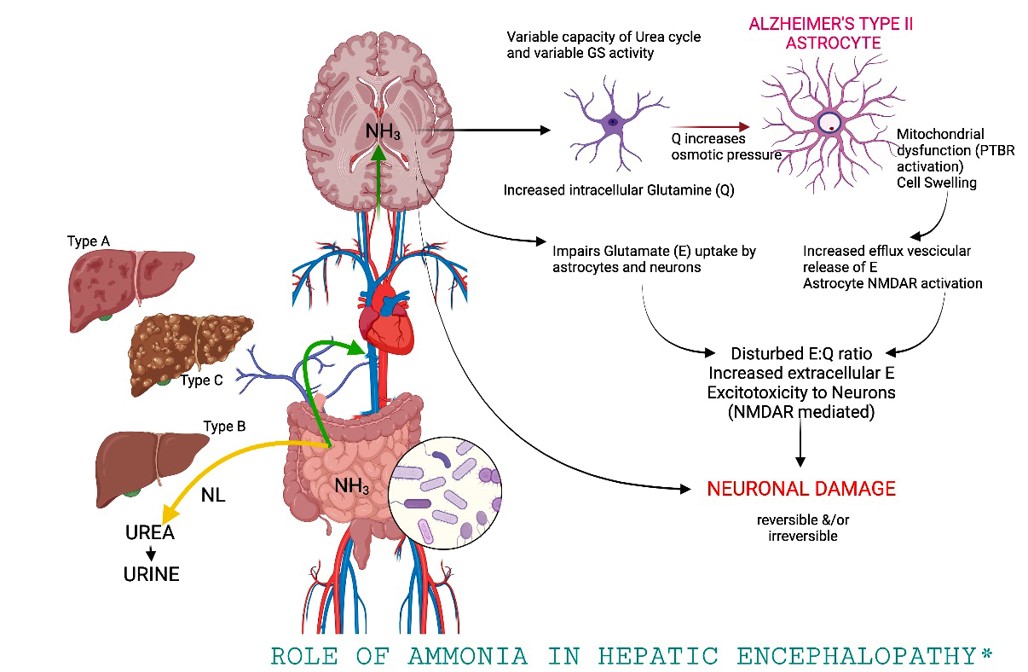
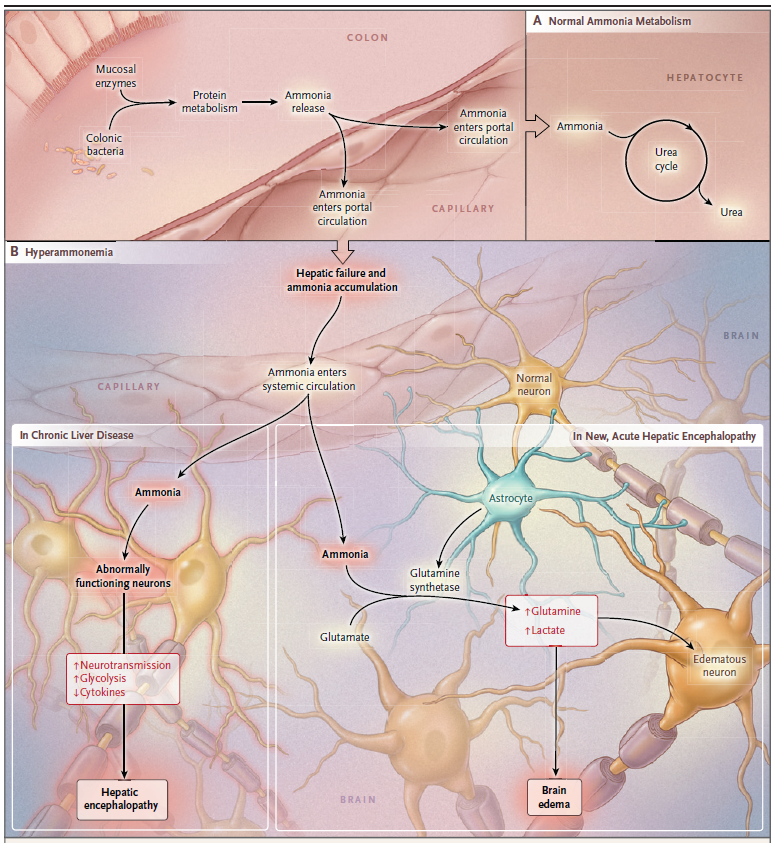
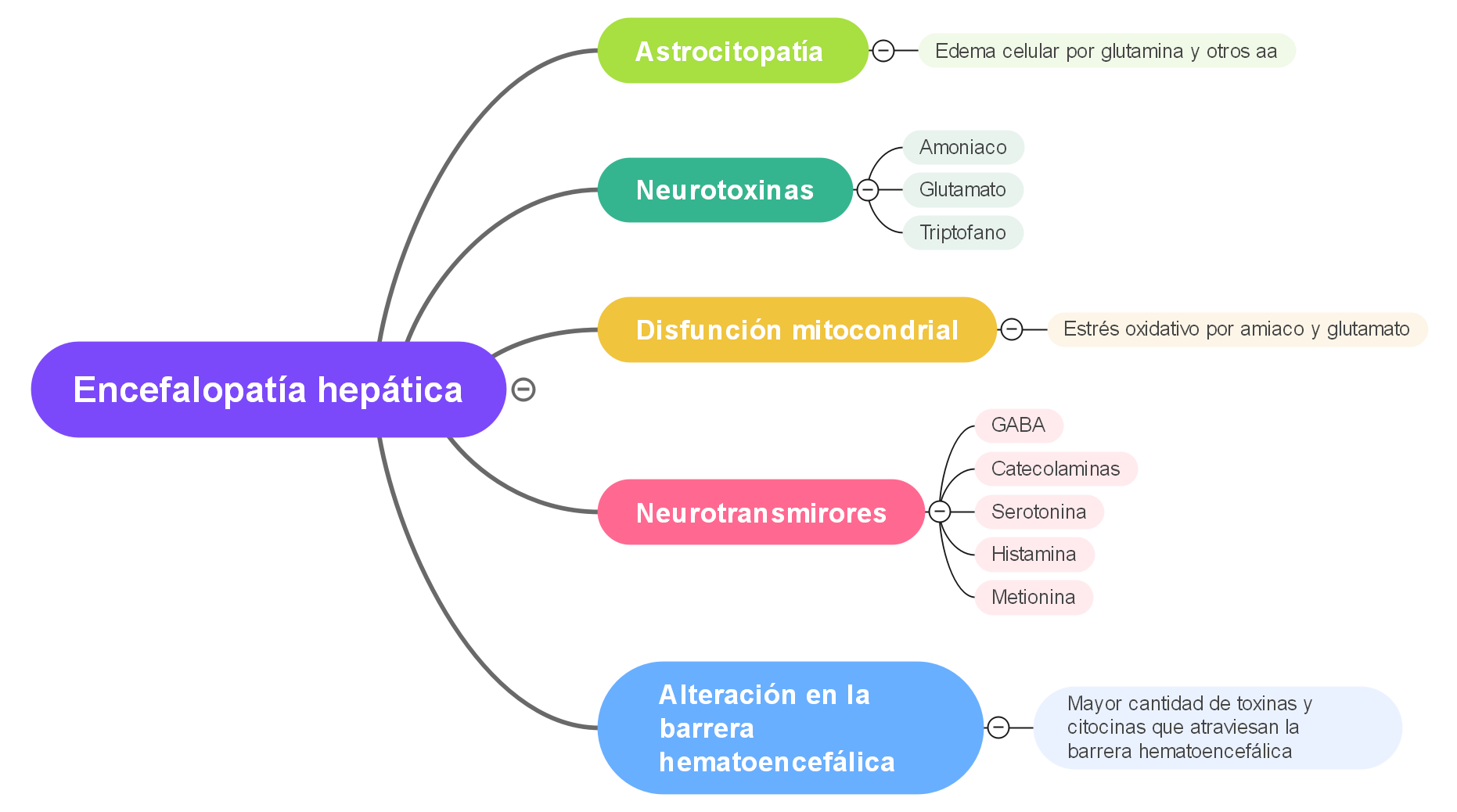
Amoníaco: el amoníaco es la neurotoxina mejor caracterizada que precipita la encefalopatía hepática. El tracto gastrointestinal es la principal fuente de amoníaco, que entra en la circulación a través de la vena porta. El amoníaco es producido por enterocitos a partir de glutamina y por catabolismo bacteriano colónico de fuentes nitrogenadas, como la proteína ingerida y la urea secretada. El hígado intacto elimina casi todo el amoníaco de sistema porta, convirtiéndolo en glutamina y evitando la entrada en la circulación sistémica.
Sin embargo, la glutamina se metaboliza en las mitocondrias produciendo glutamato y amoníaco, los cuales interfieren con la función mitocondrial que conduce a la disfunción de los astrocitos.
El aumento del amoníaco sanguíneo en la enfermedad hepática avanzada es una consecuencia de la función hepática deteriorada y de la derivación de la sangre alrededor del hígado. El desgaste muscular, una ocurrencia común en estos pacientes, también puede contribuir ya que el músculo es un sitio importante para la eliminación extrahepática de amoníaco. Cada vez más, el microbiota intestinal se reconoce como una fuente principal de amoníaco.
La concentración arterial de amoníaco aumenta en aproximadamente el 90 por ciento de los pacientes con encefalopatía hepática. La prueba del papel patogénico del amoníaco proviene de la eficacia de las terapias dirigidas a reducir el amoníaco plasmático en la mejora de la encefalopatía hepática.
Alteración del transporte de aminoácidos en encefalopatía hepática
Hiperamonemia puede aumentar la captación cerebral de aminoácidos neutros al mejorar la actividad del transportador de L-aminoácidos en la barrera hematoencefálica. Este efecto puede ser la consecuencia del transporte de glutamina de sangre a cerebro, que se forma en exceso para la desintoxicación de amoníaco. Consistente con esta hipótesis es la observación de que el transporte de triptófano en el cerebro aumenta por las infusiones de amoníaco. La consiguiente elevación en la concentración cerebral de aminoácidos neutros tirosina, fenilalanina y triptófano puede afectar la síntesis de los neurotransmisores dopamina, norepinefrina y serotonina.
Aumento de la osmolaridad intracelular en astrocitos
El edema cerebral en hiperamonemia aguda por aumento en la osmolaridad intracelular resultante del metabolismo del amoníaco en los astrocitos para formar glutamina.
Estos datos están respaldados por mediciones in vivo en pacientes cirróticos en los que la espectroscopia de resonancia magnética de protones del cerebro mostró agotamiento de mioinositol (un signo de aumento de la osmolaridad) y aumento de la glutamina. Por lo tanto, como se mencionó anteriormente con la inserción de la derivación portosistémica intrahepática transyugular (TIPS), la derivación portosistémica agrega una contribución esencial a la patogénesis de la encefalopatía.
Además del edema celular, la vasodilatación puede contribuir al aumento de la presión intracraneal en la insuficiencia hepática aguda. La liberación de glutamato inducida por amoníaco y el aclaramiento alterado de glutamato pueden elevar los niveles de glutamato extracelular y causar sobreestimulación de los receptores de N-metil-D-aspartato (NMDA). La activación del receptor NMDA desencadena el óxido nítrico sintetasa (n-NOS) a través de un mecanismo mediado por calmodulina. La síntesis catalizada por NOS de óxido nítrico (NO) produce vasodilatación. Por lo tanto, la acumulación de agua cerebral inducida por el amoníaco (probablemente debido a edema de los astrocitos) desencadena la hiperemia cerebral.
Alteración en la actividad eléctrica neuronal
el amoníaco afecta directamente la actividad eléctrica neuronal al inhibir la generación de potenciales postsinápticos excitatorios e inhibitorios.
La encefalopatía hepática se caracteriza por alteraciones bioquímicas en las funciones asociadas con las membranas neurales, como los cambios en la captación de neurotransmisores, en las actividades enzimáticas y en la expresión de los receptores de neurotransmisores. Una posible vía común para estos cambios puede ser alteraciones en las propiedades de la membrana en la encefalopatía hepática. Se documentaron alteraciones macroscópicas en la composición lipídica de la membrana cortical cerebral (incluida una disminución del colesterol, fosfatidilserina, esfingomielina, ácidos grasos mono y poliinsaturados) y la fluidez de la membrana anular en ratas con encefalopatía hepática.
Sistema de neurotransmisores GABA-benzodiazepinas
Se ha propuesto un papel para el aumento del tono del sistema de neurotransmisores inhibidores del ácido gamma-aminobutírico (GABA) A-benzodiazepina en el desarrollo de encefalopatía hepática.
Benzodiacepinas endógenas
Las benzodiazepinas endógenas involucradas en la activación de la neurotransmisión GABAA-érgica se han aislado, caracterizado e identificado positivamente por cromatografía de gases-espectroscopia de masas como benzodiazepinas en cerebro, sueros y LCR de animales de experimentación y humanos con insuficiencia hepática aguda debido a la toxicidad del paracetamol. La concentración cerebral de estas sustancias se correlacionó estrechamente con el grado de deterioro neurológico en un modelo animal.
Neuroesteroides
Los neuroesteroides son metabolitos de la progesterona y son compuestos neuroactivos endógenos. La alopregnanolona y la tetrahidrodesoxicorticosterona son potentes moduladores alostéricos positivos selectivos del complejo receptor GABAA. La administración de estos esteroides induce efectos conductuales que incluyen sedación, una propiedad consistente con la mejora de la inhibición neuronal característica de la HE. En un informe, las concentraciones de alopregnanolona y pregnenolona (un precursor de neuroesteroides) aumentaron en los cerebros de pacientes en coma hepático.
Neurotransmisión glutamatérgica
Cada vez hay más pruebas de que las alteraciones de la función glutamatérgica están implicadas en la patogénesis de las consecuencias del sistema nervioso central de la insuficiencia hepática aguda.
Receptores de glutamato : hay tres subtipos principales de receptores de glutamato, definidos de acuerdo con su acoplamiento a los canales iónicos y su afinidad con ciertos ligandos:
- N-metil-D-aspartato (NMDA)
- No NMDA – amino-3-hidroxi-5-metil-4-isoxazol ácido propiónico (AMPA) y kainato
- Receptor metabotrópico de glutamato
La pérdida selectiva de sitios AMPA es consistente con la inhibición de la despolarización neuronal mediada por AMPA resultante de la exposición de las neuronas del hipocampo a concentraciones milimolares de amoníaco. Los sitios NMDA son exclusivamente neuronales, mientras que los sitios kainato y AMPA se localizan tanto en neuronas como en astrocitos. Por lo tanto, la pérdida selectiva de sitios no NMDA en la insuficiencia hepática aguda puede reflejar cambios astrocíticos. Debido a que los receptores astrocíticos de glutamato están implicados en la recaptación de potasio y neurotransmisores, las alteraciones en su densidad podrían resultar en una excitabilidad neuronal alterada y podrían contribuir a la disfunción neurológica característica de la HE en la insuficiencia hepática aguda.
Catecolaminas
Las concentraciones alteradas de catecolaminas en la encefalopatía hepática se han relacionado con el metabolismo alterado de los aminoácidos. En la insuficiencia hepática crónica, las concentraciones plasmáticas y cerebrales de aminoácidos aromáticos (AAA; fenilalanina, triptófano y tirosina) aumentan, mientras que las de los aminoácidos de cadena ramificada (BCAA) valina, leucina e isoleucina se reducen.
Dado que estos aminoácidos comparten un portador común en la barrera hematoencefálica, la disminución de las concentraciones de BCAA en la sangre puede resultar en un mayor transporte de AAA al cerebro. Una baja proporción molar de BCAA plasmática a AAA es un hallazgo consistente en pacientes con cirrosis y HE, pero también ocurre en pacientes sin HE. Esta relación se correlaciona estrechamente con los índices de función hepática, con una relación disminuida que implica una función hepatocelular deficiente.
Alteración en la barrera hematoencefálica
La captación cerebral de varias sustancias trazadoras aumenta en varios modelos animales de insuficiencia hepática aguda.
La permeabilidad hematoencefálica se mantuvo sin cambios en un estudio de pacientes con enfermedad hepática crónica encefalopatía hepática. Por el contrario, se han demostrado cambios específicos en el transporte de la barrera hematoencefálica en pacientes con encefalopatía hepática. De particular interés son los cambios en el transporte de aminoácidos en el cerebro. Los aminoácidos como la tirosina, la fenilalanina y el triptófano son precursores de los neurotransmisores dopamina, norepinefrina y serotonina, mientras que otros aminoácidos, como el glutamato, el aspartato, la taurina y la glicina, son neurotransmisores en sí mismos.
Alteraciones en el metabolismo energético
El suministro de energía no perturbado y el metabolismo energético son un requisito previo para la función cerebral normal. La glucosa es el combustible energético cerebral más importante y la hipoglucemia puede ocurrir en las etapas terminales de la insuficiencia hepática debido a la gluconeogénesis hepática alterada. Sin embargo, la administración de glucosa no es suficiente para normalizar la función cerebral en la encefalopatía hepática.
Mayor susceptibilidad a infecciones y a toxinas
El síndrome de respuesta inflamatoria sistémica (SRIS) resulta de la liberación y circulación de citoquinas y mediadores proinflamatorios. La encefalopatía asociada a la sepsis se caracteriza por cambios en el estado mental y la actividad motora, que van desde el delirio hasta el coma. Hasta un tercio de los pacientes con sepsis tienen un nivel reducido de conciencia, que es un factor pronóstico independiente para el aumento de la mortalidad. Las posibles causas de disfunción cerebral incluyen alteraciones en el flujo sanguíneo cerebral, metabolitos cerebrales y la liberación de mediadores inflamatorios; Es importante destacar que estos procesos ocurren sin la infección directa del tejido cerebral.
Durante un episodio de sepsis, las citoquinas no pueden difundirse a través de la barrera hematoencefálica y, por lo tanto, no pueden tener un efecto directo. Sin embargo, el sistema inmune periférico puede conducir a la producción de citoquinas proinflamatorias (tanto en la periferia como en el cerebro). Estas citoquinas proinflamatorias pueden indicar al cerebro que provoque una respuesta. La señalización cerebral puede ocurrir a través del transporte directo de la citoquina a través de la barrera hematoencefálica.
Otro factor potencial que incita una respuesta inflamatoria es la translocación bacteriana de organismos desde el intestino, lo que resulta en endotoxemia crónica. La translocación bacteriana puede activar citoquinas/quimiocinas proinflamatorias y neutrófilos a través de receptores tipo Toll y quimiocinas.
Enlace externo
Hepatic encephalopathy | Deranged Physiology
5 Pearls on Hepatic Encephalopathy | Core IM Podcast
Pathology Outlines – Hepatic encephalopathy
Enlace interno
Gastroenterología » Medical & Gabeents
ARCHIVOS



