
 Gabe
GabeLa esclerosis lateral amiotrófica (ELA), anteriormente conocida como enfermedad de Lou Gehrig, es una enfermedad neurodegenerativa con disfunción de la neurona motora superior e inferior. La enfermedad se manifiesta más comúnmente entre los cincuenta y setenta años de edad, a menudo comenzando con debilidad asimétrica en las manos o los pies. Sin embargo, la presentación inicial es muy variable y algunos pacientes presentan síntomas atípicos/inespecíficos, como cambios vocales sutiles.
A medida que la enfermedad progresa, la mayoría de los pacientes eventualmente desarrollan uno o ambos de los síntomas potencialmente mortales: insuficiencia respiratoria y disfagia. El riluzol y la edaravona son actualmente los únicos fármacos aprobados para el tratamiento de la ELA.
La atención multidisciplinaria es extremadamente importante e incluye atención de enfermería, fisioterapia y, finalmente, ventilación asistida y alimentación enteral. La mayoría de los pacientes morirán dentro de 3-5 años, aunque aproximadamente el 30% tiene una probabilidad de vivir más tiempo.
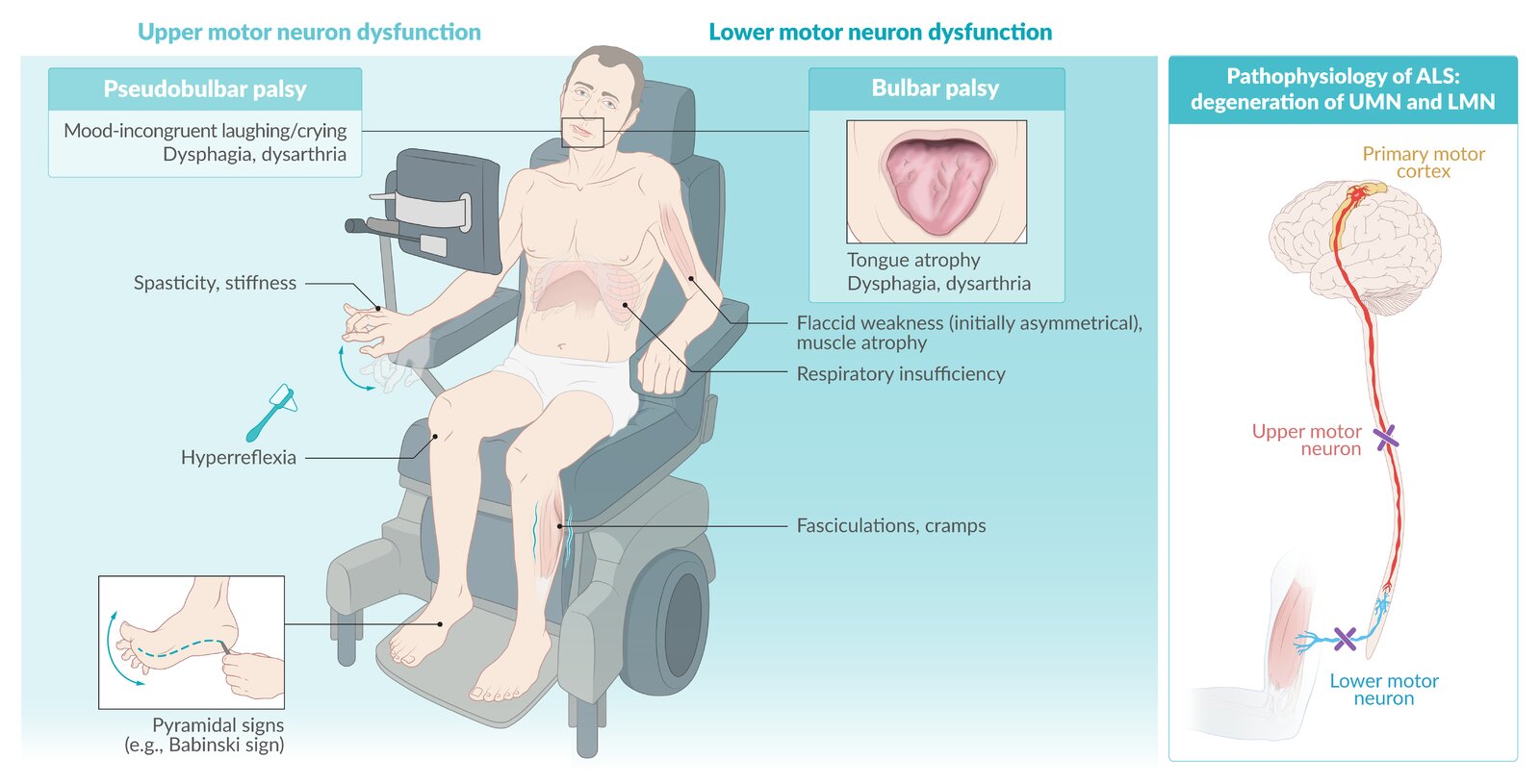
El sello clínico de la esclerosis lateral amiotrófica es la combinación de signos y síntomas de la neurona motora superior e inferior.
Los hallazgos de la neurona motora superior son el resultado de la degeneración de las neuronas motoras del lóbulo frontal y del tracto corticoespinal.
Los hallazgos de las neuronas motoras inferiores son una consecuencia directa de la degeneración de las neuronas motoras inferiores en el tronco encefálico y la médula espinal.
Epidemiologia
- Prevalencia: 5/100.000 habitantes en los EE.UU.
- Incidencia: 2-3 casos/100.000 habitantes por año en todo el mundo
- Sexo: ♂ > ♀
- La edad media de aparición es de 65 años.
- Antecedentes familiares de ELA en el 5-10% de los casos; 90-95% son esporádicos
Etiología
La causa definitiva de la ELA aún se desconoce. Los estudios han sugerido una interacción entre la predisposición genética y los factores ambientales.
Genética
Se han encontrado mutaciones de los siguientes genes en aproximadamente 70% casos familiares y algunos casos esporádicos:
- SOD1; Códigos para la superóxido dismutasa
- TARDBP; Сodes para la proteína
- TDP-43 implicada en la reparación del ADN En la ELA
- TDP-43 forma inclusiones dentro de las neuronas motoras. Representa ∼ 5% de los casos familiares de ELA.
- C9orf72; Gen mutado más común en la ELA familiar (representan el 30-40% de los casos de ELA familiar) Asociado con una combinación de ELA y demencia frontotemporal
- FUS Las mutaciones se asocian con una ELA de inicio joven que progresa rápidamente. Representa ∼ 5% de los casos familiares de ELA.
Factores de riesgo ambientales:
- β-N-metilamino-L-alanina
- Plaguicidas (por ejemplo, cis-clordano, pentaclorobenceno)
- Traumatismo craneoencefálico
- Tabaquismo
Patología
Clásicamente afecta a todo el sistema de neuronas motoras en dos o más niveles (degeneración de la neurona motora superior e inferior).
- Neuronas motoras superiores en el giro precentral y, con frecuencia, en la corteza prefrontal.
- Neuronas motoras inferiores en el asta anterior de la médula espinal y el tronco encefálico.
Los posibles mecanismos subyacentes incluyen procesamiento anormal de ARN y agregación de proteínas, excitotoxicidad, disfunción mitocondrial y neurofilamentos defectuosos.
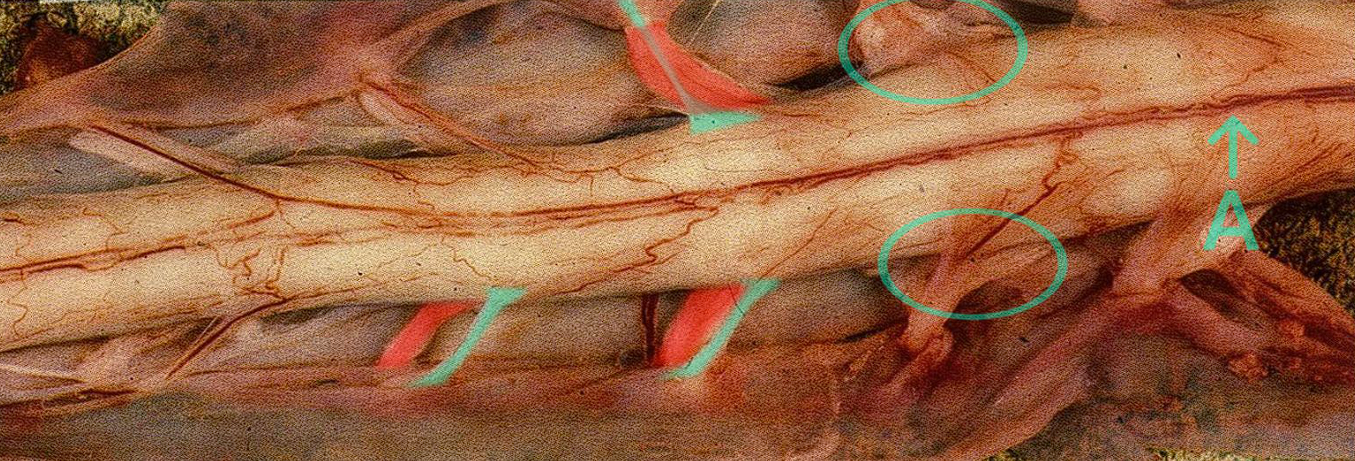
Médula espinal de un paciente con esclerosis lateral amiotrófica (muestra patológica, vista ventral) que muestra una degeneración relativa de las raíces ventrales (marcas verdes) en comparación con las raíces dorsales (marcas rojas). La atrofia de algunas de las raíces ventrales es tan grave que apenas se distinguen de la propia médula espinal (marcas ovaladas). (A = arteria espinal anterior)
Espectro de trastorno clínico
ELA es la forma más común de enfermedad de la neurona motora e incluye patología de la neurona motora superior e inferior. El espectro de la enfermedad de la neurona motora también incluye otras afecciones que pueden ser variantes de la ELA o pueden representar diferentes patrones de evolución en la ELA. Estos incluyen:
- Atrofia muscular progresiva
- Esclerosis lateral primaria
- Parálisis bulbar progresiva
- Síndromes de brazo y pierna de inestable
Síntomas
Los síntomas y signos clínicos primarios de ELA pueden producir deterioro que afecta la función de las extremidades, bulbar, axial y respiratoria.
Los síntomas de la neurona motora superior resultan en lentitud de movimiento, incoordinación y rigidez típicamente con relativamente poca debilidad.
Los síntomas de la neurona motora inferior causan debilidad, generalmente acompañada de atrofia y fasciculaciones. Los espasmos musculares también son comunes.
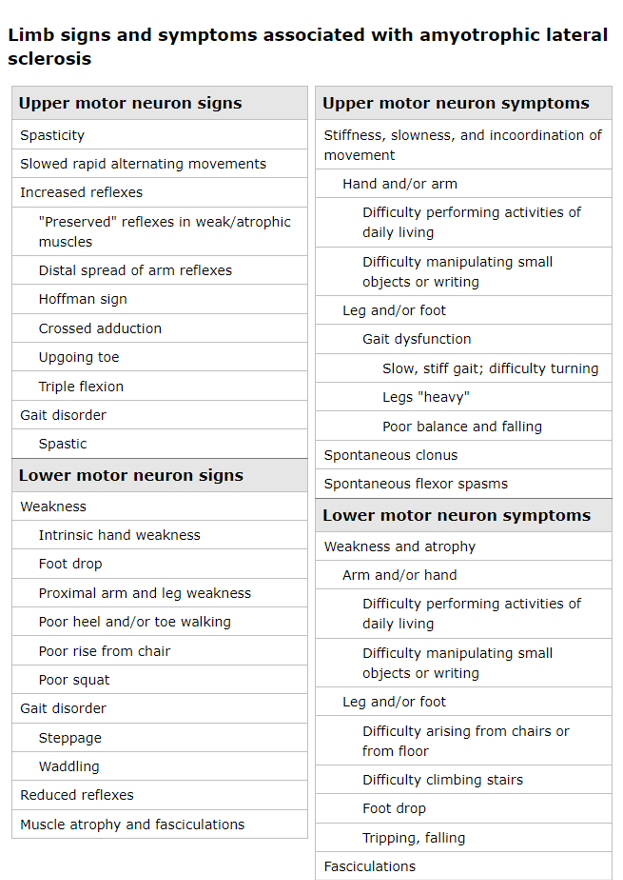
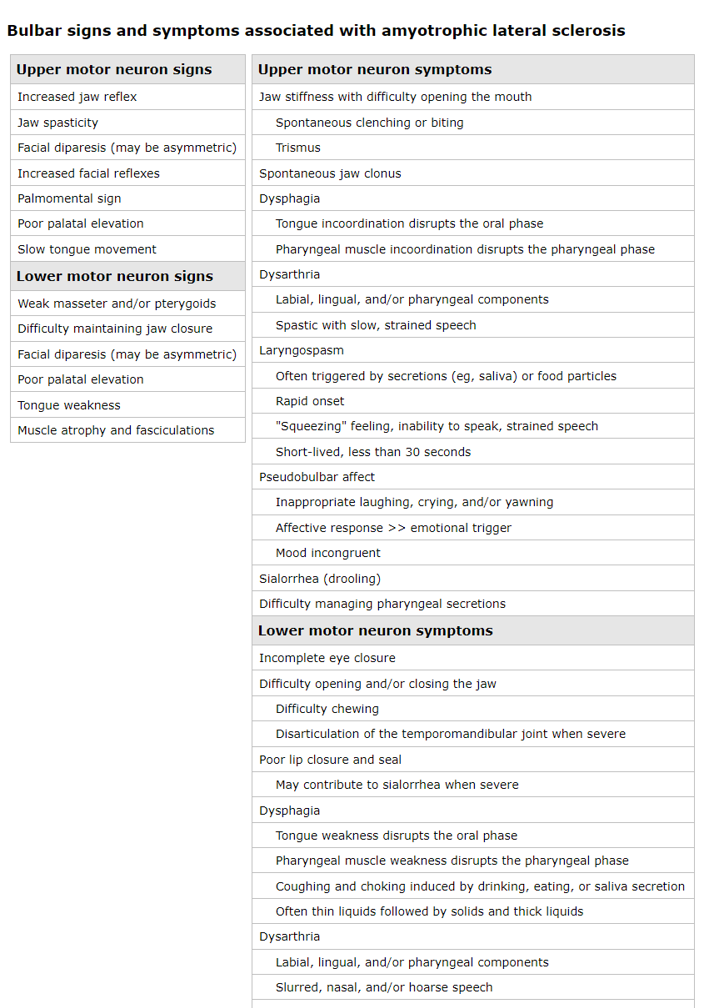
El deterioro cognitivo
Típicamente relacionado con la disfunción ejecutiva frontotemporal, puede preceder o seguir la aparición de la disfunción de la neurona motora superior y a la disfunción de la neurona motora inferior en pacientes con ELA. La demencia frontotemporal puede estar asociada con ELA en 15 a 50 % de los casos.
Los síntomas autonómicos pueden ocurrir en la ELA a medida que la enfermedad progresa. El estreñimiento ocurre con frecuencia y es probable que sea multifactorial. La disfagia para líquidos delgados relacionados con la debilidad del músculo faríngeo puede conducir a la deshidratación, lo que puede exacerbar el estreñimiento.
Los síntomas extrapiramidales y los signos de parkinsonismo pueden preceder o seguir a los síntomas de la neurona motora superior e inferior. Estas características extrapiramidales pueden incluir enmascaramiento facial, temblor, bradicinesia e inestabilidad postural.
El deterioro sensorial puede ocurrir en 20 a 30 % de los pacientes con ELA, pero el examen sensorial suele ser normal. El dolor nociceptivo en la ELA puede surgir de una variedad de causas que incluyen movilidad reducida, calambres musculares, espasticidad muscular y afecciones comórbidas.
Características clínicas clave
Las manifestaciones clínicas de la esclerosis lateral amiotrófica (ELA) incluyen la presencia de signos de neuronas motoras superiores e inferiores, progresión de la enfermedad y la ausencia de una explicación alternativa.
Diagnóstico
El diagnóstico de ELA se realiza en pacientes que cumplen con los criterios diagnósticos evaluados por la historia y el examen físico, respaldados por estudios de electrodiagnóstico y no excluidos por neuroimagen y estudios de laboratorio.
Los criterios utilizados para el diagnóstico de la ELA han evolucionado con el tiempo. Los criterios revisados El Escorial World Federation of Neurology criteria, también conocidos como los criterios de Airlie House, incluyen signos de neuronas motoras superiores e inferiores y progresión de los síntomas a lo largo del tiempo. Criterios de Gold Coast simplifican los criterios revisados de El Escorial para la ELA.
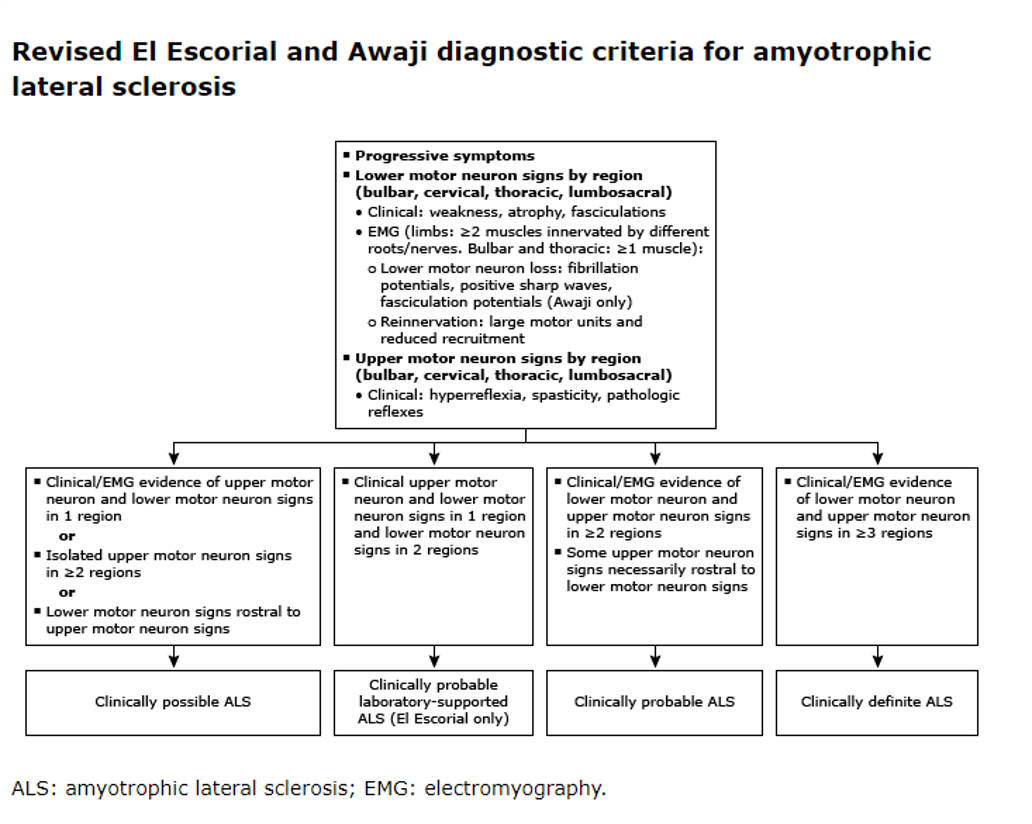
Estudios electrodiagnósticos
Se recomienda realizar pruebas de electrodiagnóstico con estudios de conducción nerviosa y electromiografía (EMG) en todos los pacientes con sospecha de ELA. Los estudios de conducción nerviosa sensorial y motora suelen ser normales en la ELA, aunque las amplitudes del potencial de acción muscular compuesto (CMAP) pueden reducirse en los músculos gravemente atróficos y denervados. La EMG generalmente revela características combinadas de denervación aguda y crónica en la ELA.
Pruebas diagnósticas adicionales:
Se recomienda realizar TC e IMR de encéfalo y pruebas de laboratorio de rutina en todos los pacientes con sospecha de ELA para excluir diagnósticos alternativos. Otras pruebas de diagnóstico se realizan para pacientes seleccionados.
Neuroimagen
La evaluación por resonancia magnética (IRM) debe incluir todos los segmentos rostral a los hallazgos clínicos; Esto incluye el cerebro, la columna cervical y la columna torácica cuando los hallazgos de la neurona motora superior están en las piernas. La resonancia magnética convencional suele ser normal en la ELA.
Pruebas de laboratorio
Las pruebas de rutina de sangre y orina se utilizan para todos los pacientes para excluir diagnósticos alternativos. La punción lumbar para el análisis del líquido cefalorraquídeo se realiza si existe sospecha clínica para el diagnóstico de polineuropatía desmielinizante inflamatoria crónica, enfermedad de Lyme, infección por VIH o linfoma o si hay un síndrome de neurona motora inferior aislada rápidamente progresiva. Es posible que se requieran pruebas de laboratorio adicionales en ciertos entornos clínicos.
Otras pruebas para algunos pacientes
Las pruebas genéticas no son una parte requerida de la evaluación diagnóstica en la ELA, pero pueden ser útiles para hacer el diagnóstico en la ELA familiar, que representa aproximadamente el 10 % de todos los casos de ELA.
La biopsia muscular no es una parte rutinaria de la evaluación diagnóstica de la ELA, pero debe realizarse si existe sospecha clínica de miopatía inflamatoria.
Diagnóstico diferencial
El diagnóstico diferencial de la ELA es extenso. Esto incluye neuropatía motora multifocal, radiculomielopatía cervical, fasciculaciones benignas, miopatías inflamatorias, síndrome post-polio, amiotrofia monomélica, paraplejia espástica hereditaria, atrofia muscular espinobulbar, miastenia gravis e hipertiroidismo.
Progresión clínica
ELA es un trastorno implacablemente progresivo con un curso clínico que generalmente es lineal. Los síntomas generalmente se propagan dentro del segmento de inicio y luego a otras regiones en un patrón relativamente predecible. El curso progresivo de la ELA eventualmente produce insuficiencia respiratoria neuromuscular potencialmente mortal.
Enlace interno
Neurología » Medical & Gabeents
Referencia
Hardiman, O., Al-Chalabi, A., Chio, A. et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers 3, 17071 (2017). https://doi.org/10.1038/nrdp.2017.71
Enlace externo



